
A highly expandable and tough polyacrylamide – alginate microcapsule†
Yan-Li Li,
Ming-Lu Zhu,
Xiao-Yu Li,
Xiao-Heng Li and
Yong Jiang *
*
School of Chemistry and Chemical Engineering, Southeast University, Jiangning, Nanjing, Jiangsu 211189, P. R. China. E-mail: yj@seu.edu.cn
First published on 20th April 2016
Abstract
A highly expandable and tough microcapsule was prepared by water-in-oil emulsion polymerization. The diameter of the microcapsule could be expanded to about 75 times larger than the original size without breakage. Furthermore, this capsule was pH stimuli-responsive, and might be used in materials or biotechnology applications.
Recent advances in medicine and biotechnology have prompted the need to develop nano-engineered encapsulation-delivery systems that can encapsulate a wide variety of therapeutics or molecules. These systems should be “intelligent” enough to deliver their payloads at a well-defined time, place, or after a specific stimulus without causing any damage.1 Various methods have been developed to prepare polymer based capsules such as self-assembly,2–6 layer-by-layer deposition,7–15 in situ polymerization16–20 and precipitation.21–23 However, the mechanical strength of the capsule shell, which is one of the most important properties, hinder the development of their application. Many efforts were made to produce robust and stretchable capsules by numerous techniques such as template method,18 interfacial reaction of polymers24,25 and layer-by-layer assembly.26,27 Different kinds of microcapsules were also prepared in our group.28,29 Nevertheless, it is still difficult to get a microcapsule which is both stretchable and robust.
In the past, synthetic as well as natural materials were used for encapsulation-delivery systems.1,8,10,13,18,29 Polyacrylic acid, polyacrylamide (PAAm), poly-hydroxyethyl methacrylate, polycaprolactone and glycolide were commonly used synthetic polymers, whereas chitosan, alginate and gelatin were the most frequently used biopolymers. For the development of a stretchable and robust microcapsule, alginate was selected as one of the components because of its favourable properties such as good biocompatible, wide availability, low cost and simple gelling procedure under mild conditions.30,31 The gelation process of alginate is driven by the interactions of G and MG blocks with Ca2+ to form strong and weak junctions.30 PAAm was selected as a second component to make microcapsules because of its previous applications in drug delivery application, contact lenses and biosensor preparation.32–35
Now in this paper, a highly expandable and tough microcapsule was prepared by water-in-oil emulsion polymerization, in which alginate and PAAm triple networks36 were employed as microcapsule shell and CaCO3 microsphere was used as a template. The most important finding was that the diameter of the microcapsule could be expanded to about 75 times larger than the original size without breakage (i.e. the volume increased to 4.2 × 105 times). Besides, this capsule showed pH stimuli-responsive properties. When exposed to acid, the capsule was expanded and released the preloaded molecules. These unique features of this microcapsule made it useful for molecule encapsulation, release and other repair applications that might need such expandable materials for retreatment.
Fig. 1 illustrates the strategy for preparing the highly stretchable and tough microcapsule. Please see Experimental section in the ESI† for the detailed preparation process. First, CaCO3 microsphere, AAm and alginate were mixed together in deionized water to form water phase. Since CaCO3 microspheres were positively charged at this suspending medium (pH < 8.5),37 the negative charged alginate was absorbed on their surface by electrostatic interactions in a way like the absorption of polyelectrolyte to CaCO3 particles during layer-by-layer assembling.28,37 Then, the mixture was added into prepared oil phase to form a water-in-oil system. As the amount of CaCO3 particles and water phase were controlled in a well-designed ratio, water phase mainly existed as a thin layer around each CaCO3 particle and there was almost only one CaCO3 particle in each water drop. When potassium peroxydisulfate was added as radical initiator, AAm began to be crosslinked by N,N-methylene bisacrylamide (MBA) thus formed PAAm network via inversed emulsion polymerization.38 After the polymerization, PAAm layer was synthesized around CaCO3 particle and alginate was dispersed in PAAm layer by the interaction between amine group of acrylamide and carboxyl group of alginate.36 Model molecules doxorubicin hydrochloride (Dox) could be loaded in the capsules after the polymerization or it could also be preloaded during the preparation of CaCO3 particles.
 | ||
| Fig. 1 Schematic drawing of the preparation process of crosslinked alginate–PAAm hollow capsules. (a) CaCO3 microspheres were dispersed in aqueous solution of alginate and acrylamide (AAm). (b) Alginate was adsorbed on the surface of CaCO3 microsphere by charge interaction in acrylamide solution. (c) Water phase was added into oil (n-heptane) phase to produce a water-in-oil emulsion and water phase mainly existed as thin layer around CaCO3 cores and there was almost only one CaCO3 particle in each water drop. (d) After polymerization, PAAm network was synthesised around each CaCO3 microsphere. (e) CaCO3 core was decomposed by acid. At the same time, Ca2+ began to crosslink alginate to form alginate network and CO2 began to expand microcapsules. (f) With the continue release of Ca2+ and CO2, capsules became larger and larger. | ||
When acid was added to the emulsion, CaCO3 core began to decompose inside the capsule. The released Ca2+ began to crosslink the alginate and strengthen the capsule shell. At the same time, the produced CO2 tended to blow the microcapsule and push the preloaded molecules out of the capsule. Then, the capsule became larger and larger as reaction between CaCO3 and acid kept on. Finally, after CaCO3 microspheres decomposed completely, final diameters of capsules became about 75 times bigger than their initial sizes, which meant the volume of microcapsule could increase about 4.2 × 105 times.
Using CaCO3 microsphere as template to prepare microcapsule is an old and traditional method to make capsules.39,40 However, in this paper, this old and simple method was designed in a very smart way. Here, CaCO3 microspheres played at least four important roles during preparations of alginate–PAAm capsules. First, they worked as sacrificial microcapsule templates for the covalent crosslinking of PAAm network on their surface. Second, when they were decomposed by acid, the released Ca2+ was used for the ionic crosslinking of alginate network. Third, the CO2 gas produced by the decomposition of CaCO3 was used to blow microcapsules and tested stretchable properties of capsules. Fourth, CaCO3 microsphere could be used as vector to load drug or other molecules and preloaded molecules can be driven out by the generated CO2 gas.
To confirm the alginate–PAAm capsule was synthesized properly, the prepared capsule was measured by FTIR and results were shown in (ESI) as Fig. S1.† A new peak at 1383 cm−1 for C–N stretching of secondary amide was found in the spectrum of the capsule compared with that of alginate and PAAm. Furthermore, the intensities of primary amide peaks (1636 cm−1 and 1460 cm−1) and NH2 in-plane rocking peak (1124 cm−1) increased, as well as the intensities of O–H stretching peak (3450 cm−1) and symmetric C–O stretching (1090 cm−1). All these results indicated that new bonds were formed between amino groups of PAAm and carboxyl groups of alginate.
CaCO3 microsphere was prepared using the former methods.28 It had rough surface as shown by the SEM image in Fig. 2a. Additional SEM image of CaCO3 microspheres was presented in Fig. S2a† and their size distribution was shown in Fig. S2b in ESI.† The prepared CaCO3 microspheres had a narrow size distribution of 3.2 ± 1.1 μm. Microcapsule layer was formed on the CaCO3 microsphere after inverse emulsion polymerization of AAm. Surface morphology of the capsule was shown in Fig. 2b, as well as Fig. S3a and b in ESI.† Semi-transparent and smoothly PAAm gel layer was seen clearly on the surface of CaCO3 microsphere as shown in Fig. 2b. In Fig. S3a and b,† more PAAm capsules were found and some of them were ruptured, which might be caused by long time irradiation of high energy electron beam in vacuum during the SEM measurements. Just seeing from SEM images of capsules, their sizes were quite uniform with diameters about 3.7 ± 0.7 μm. The diameter of the capsule was about 0.5 μm bigger than that of CaCO3 microsphere, which should be the thickness of the PAAm–alginate hydrogel layer. Fig. 2c was the ultrathin cross section image of the capsule measured by TEM and higher magnification image could be seen in Fig. S3c.† Loose structure of CaCO3 microsphere could be seen with a quite big free room inside. So it is a great advantage to use the free volume to load drug or other molecules for next-step controlled release experiments.
 | ||
| Fig. 2 SEM and TEM images show the morphologies of the CaCO3 microsphere, microcapsule and hollow capsules. (a) SEM image of CaCO3 microsphere. (b) SEM image of alginate–PAAm microcapsule with CaCO3 microsphere inside. (c) TEM image of ultrathin slices of microcapsule with CaCO3 microsphere inside. (d) SEM image of hollow capsules after the CaCO3 core was decomposed by acid. (e) SEM images of cross section of hollow capsules after the CaCO3 core was decomposed by acid. (f) SEM images of capsules after the CaCO3 core was removed by EDTA. | ||
When acid was added into the microcapsule emulsion, CaCO3 core began to decompose. As a result of it, the microcapsule turned into big hollow capsule without breakage. The blown capsules were shown in Fig. 2d and diameters had a relative wide distribution from about 80 μm to 350 μm. The cross section image of the blown capsules could be seen in Fig. 2e. Ultrathin shell of blown capsules exhibited uniform thickness. However, when using EDTA to decompose the CaCO3 microsphere, the size of microcapsules showed unapparent change and the SEM image is shown in Fig. 2f. The hollow capsules looked like sea urchins and some of them were broken, which might be caused by long-time irradiation of electron beam in vacuum (the same reason with that of ruptured microcapsule).
Optical microscope was adopted to observe the dynamic expansion process of alginate–PAAm capsules and the results were shown in Fig. 3. In the beginning, the original diameters of the microcapsules were about 3.7 ± 0.7 μm without adding acid (Fig. 3a). After acid was added for 10 min, the diameters of the microcapsules increased to about 28 ± 7 μm (Fig. 3b). And the diameters of the capsules further increased to 101 ± 34 μm and 150 ± 39 μm after 2 h and 8 h of incubation, respectively (Fig. 3c and d). At last, after 24 h of treatment, the diameters of the capsules almost reached their maximum values of about 276 ± 108 μm. The relationship between the diameters of the capsules and incubation time was shown in Fig. 3f. The diameters of the microcapsules can be expanded to about 75 times (75 = 276/3.7, calculated by average diameters) to its original diameters without fracture according to the observation using optical microscope.
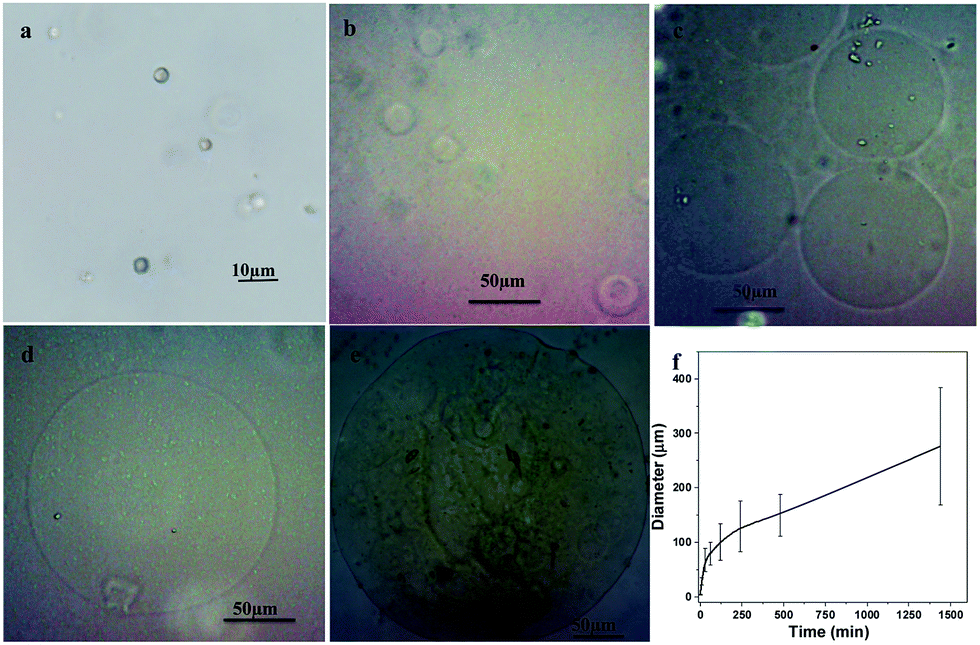 | ||
| Fig. 3 Optical microscope images show the dynamic size changes of alginate–PAAm capsules when acid was added. (a) Original microcapsules without adding acid. The microcapsule turned into big capsules after acid was added for (b) 10 min, (c) 2 h, (d) 8 h and (e) 24 h. (f) The relationship between capsule sizes and incubation time after adding acid. | ||
The expanding process of capsules can be taken as swelling of gel (capsule shell) forced by produced CO2 gas. According to the swelling kinetics of gels proposed previously, most theories were based on equilibrium of the force or the free energy of gel and fluid. No matter the gel was taken as linear elasticity41,42 or the force on the gel was defined as the functional derivative of the free energy function,43 the gel must reach a stationary state in equilibrium with the surrounding fluid.41,44–49 Here, in our work, the swelling of this composite gel network should follow the same swelling theory proposed previously. To reach an equilibrium state, the capsule shell must make some change (swelling and expansion) to resist the swelling force from solution and blow force from CO2. The expansion process of capsules could be divided into two parts, one was the swelling of the capsule shell (gel network) by adsorbing acid solution and the other was the swelling of the capsule cavity caused by blow force came from the produced CO2 gas. As CaCO3 could react with acid quickly, a lot of CO2 gas was produced, which resulted in quick expansion happened in the beginning. On the contrary, the size change of the capsules was not obvious when CaCO3 core was decomposed by EDTA (Fig. 2f). It is because no CO2 gas was produced to expand the capsule shell during this process.
As for the capsule shell, triple crosslinking networks were formed inside the composition gel. They were covalent crosslinks of PAAm network through polymerization of AAm by MBA, ion crosslinks of alginate network after Ca2+ crosslinking alginate and alginate/PAAm hybrid network. It was the interaction of three networks that protected the capsule shell (gel) from breaking. When capsules were immersed in pH 1.2 (<pKa of alginate), part of alginate–Ca2+ network ruptured as MG blocks didn't contribute to alginate–Ca2+ junction in acid condition.30,31,50 This rupture process protected covalent bonds in PAAm network from damaging and preserved the basic shape of capsule. The changes of alginate–Ca2+ network made these capsules highly stretchable. At the same time, the existence of crosslink in PAAm network and interaction between alginate and PAAm made the capsules still tough.
When the capsule was used as vector to transport molecules, model molecule doxorubicin hydrochloride (Dox) was loaded into microcapsule by the method used in our previous study.28,29 Please see Experimental section in the ESI† for the detailed preparation process. The released model molecule was measured by UV-Vis spectroscopy and the results were shown in Fig. 4 and S4 in ESI.† Fig. S4a† and the black line in Fig. 4 present the release of model molecule triggered by adding acid to adjust pH value to 1.2. The amount of released model molecules increased with time and reached the maximum value after about 78 h of incubation. About 90% model molecules loaded in microcapsules were released at that period of time. Fig. S4b† and the red line in Fig. 4 show the release behaviour of model molecules in PBS buffer (pH 7.4) as control. The total amount of released molecules was negligible (less than 3%) at this condition.
 | ||
| Fig. 4 Accumulative release curves as the function of incubation time at different conditions. The black line represents the release of model molecule was triggered by adjusting pH to 1.2. The blue line represents the model molecules released when using EDTA to decompose CaCO3 core. The pink line is the release curve of molecules which was first treated by EDTA for 78 h (blue line) and then immersed in acid (pH 1.2). The red line shows the molecule release of capsules in pH 7.4 solution. | ||
Besides, in order to better understand the release mechanism of molecule release in pH 1.2, EDTA was used to trigger the release as well (Fig. S4c†). The amount of model molecules released by adding EDTA came to the maximum value (10% of Dox, the blue line in Fig. 4) after EDTA was added for 0.5 hours and the amount did not change since then. It meant that about 90% of model molecules were still stored inside the microcapsule. So after microcapsules were treated by EDTA for 78 h, acid was added to adjust pH value to 1.2. The pink line in Fig. 4 shows that the remained molecules continued to quickly release again and reached the maximum value (∼65%) after about another 24 hours. Fig. 4 shows the accumulative release curves as the function of incubation time at different conditions. Fig. S5 in ESI† present the final solutions that contained all the release Dox at different conditions. The total released amount of Dox could be determined simply by their colour.
The release behaviours shown in Fig. 4 implied that the controlled release behaviours of model molecule in acid solution (pH 1.2) might be triggered by the reaction between acid and CaCO3. At pH 1.2, the junction between Ca2+ and alginate was formed when Ca2+ released by acid from CaCO3. However, this junction was weakened as MG blocks didn't contribute to alginate–Ca2+ junction in acid condition. This change could make the gel of capsule shell looser and easier to be expanded. At the same time, the capsule shell became thinner when the capsule was blown up. All these variation in capsule shells resulted in easier release of model molecules in pH 1.2.
The release of model molecules was fast at first 2 hours when the capsule shells were expanded about 30 times. The release profile shared the similar regulation with expansion process and this could well explain the mechanism of the release of model molecule in pH 1.2. The reason might be that CO2 gas produced by the reaction between acid and CaCO3 drove the model molecules out of the capsule quickly and completely.
Besides, only 10% of model molecules can be released when triggered by EDTA though EDTA could decompose the CaCO3 core. During this process, the loaded molecules loaded in CaCO3 cavity entered into capsule cavity as CaCO3 core decomposed. While, the capsule shell was not expanded because no gas produced in this situation. When the model molecules were released to solution, they need to pass the thick capsule shell without push from gas. While in PBS solution (pH 7.4) as control, CaCO3 core couldn't be decomposed in this condition. There was no change in capsule shell as well. Most of the loaded model molecules might stay inside the cavities of CaCO3 core. Only part of model molecules could be released by the osmotic pressure. Thus resulted in the little amount of released molecules (about 3%, red line in Fig. 4) as time went on.
Once the capsule synthesized by EDTA was immersed in pH 1.2 solution, the model molecule began to be released again (Fig. 4, pink line). This might be ascribed to the CO2 gas produced by the reaction between CO32− (came from the decomposition of CaCO3 with EDTA) and acid. The release of model molecules triggered by EDTA showed the importance of the change of capsule shell structure and produced gas to great release of capsules. It gave a great help to understand the release in pH 1.2. A schematic drawing of the possible mechanism of molecules released from the capsules can be seen in Fig. S6 in ESI.†
Moreover, the drug release behaviour driven by acid was similar to traditional gas-formation-based drug delivery system, which normally was prepared through complicated methods with complex structures. For example, the liquefied gas was used as a driving force for the drug delivery process.51 The interaction between effervescent layer and gas-entrapped polymeric membrane was used to control the release of drug loaded in the core pellet.52–54 Osmotic pump tablet was also prepared to achieve gas formation based controlled drug release.55 When this microcapsule was taken as a gas formation based system, CaCO3 played the role of drugs loaded tablet and the effervescent layer at the same time, and the highly stretchable and tough capsule shell played the part of gas-entrapped polymeric membrane.
In addition, the big free room in the microspheres (Fig. 2c) offered space for molecules that might be loaded in it. Molecules loaded in microcapsule were protected well by microcapsule with negligible release in neutral condition. Model molecules loaded in microcapsule showed continued release when microcapsule touched acid solution. Although this release characteristic was different with drug delivery system for oral release, it might have some other applications in the field which need pH controlled release.
Conclusions
In summary, the important characteristic of microcapsules that synthesized was its tough and highly expandability. The diameter of the microcapsule could be expanded to about 75 times larger than the original size without breakage i.e. the volume increased to 4.2 × 105 times. When the microcapsule was used as a molecule vector, it showed excellent pH sensitive molecule protection and release profiles. The molecule release driven by acid at pH 1.2 showed the similar profile as the gas-formation-based drug delivery system. These characteristics of microcapsules might be used in fields that need expandable materials to offer protection or pH sensitive controlled release of some molecules for therapy.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) with grant number 21174029, the Industry Academia Cooperation Innovation Fund of Jiangsu Province with grant number BY2014127-07 and the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).Notes and references
- L. J. De Cock, S. De Koker, B. G. De Geest, J. Grooten, C. Vervaet, J. P. Remon, G. B. Sukhorukov and M. N. Antipina, Angew. Chem., Int. Ed., 2010, 49, 6954 CrossRef CAS PubMed.
- D. E. Bergbreiter, Angew. Chem., Int. Ed., 1999, 38, 2870 CrossRef CAS.
- M. M. Conn and J. Rebek, Chem. Rev., 1997, 97, 1647 CrossRef CAS PubMed.
- J. M. Kang and J. Rebek, Nature, 1996, 382, 239 CrossRef CAS PubMed.
- J. M. Kang and J. Rebek, Nature, 1997, 385, 50 CrossRef CAS PubMed.
- P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697 CrossRef CAS PubMed.
- A. L. Becker, A. P. R. Johnston and F. Caruso, Small, 2010, 6, 1836 CAS.
- G. Berth, A. Voigt, H. Dautzenberg, E. Donath and H. Mohwald, Biomacromolecules, 2002, 3, 579 CrossRef CAS PubMed.
- F. Caruso, D. Trau, H. Mohwald and R. Renneberg, Langmuir, 2000, 16, 1485 CrossRef CAS.
- C. Gao, E. Donath, S. Moya, V. Dudnik and H. Mohwald, Eur. Phys. J. E: Soft Matter Biol. Phys., 2001, 5, 21 CrossRef CAS.
- A. P. R. Johnston, C. Cortez, A. S. Angelatos and F. Caruso, Curr. Opin. Colloid Interface Sci., 2006, 11, 203 CrossRef CAS.
- T. Shutava, M. Prouty, D. Kommireddy and Y. Lvov, Macromolecules, 2005, 38, 2850 CrossRef CAS.
- S. A. Sukhishvili, Curr. Opin. Colloid Interface Sci., 2005, 10, 37 CrossRef CAS.
- C. H. Ye, Z. A. Combs, R. Calabrese, H. Q. Dai, D. L. Kaplan and V. V. Tsukruk, Small, 2014, 10, 5087 CAS.
- W. Xu, P. A. Ledin, F. A. Plamper, C. V. Synatschke, A. H. E. Muller and V. V. Tsukruk, Macromolecules, 2014, 47, 7858 CrossRef CAS.
- M. M. Ali and H. D. H. Stover, Macromolecules, 2003, 36, 1793 CrossRef CAS.
- S. A. F. Bon, S. Cauvin and P. J. Colver, Soft Matter, 2007, 3, 194 RSC.
- A. Postma, Y. Yan, Y. J. Wang, A. N. Zelikin, E. Tjipto and F. Caruso, Chem. Mater., 2009, 21, 3042 CrossRef CAS.
- E. Quevedo, J. Steinbacher and D. T. McQuade, J. Am. Chem. Soc., 2005, 127, 10498 CrossRef CAS PubMed.
- C. Scott, D. Wu, C. C. Ho and C. C. Co, J. Am. Chem. Soc., 2005, 127, 4160 CrossRef CAS PubMed.
- P. Erni, G. Dardelle, M. Sillick, K. Wong, P. Beaussoubre and W. Fieber, Angew. Chem., Int. Ed., 2013, 52, 10334 CrossRef CAS PubMed.
- Y. J. Zhao, H. C. Shum, L. L. A. Adams, B. J. Sun, C. Holtze, Z. Z. Gu and D. A. Weitz, Langmuir, 2011, 27, 13988 CrossRef CAS PubMed.
- D. Lensen, D. M. Vriezema and J. C. M. van Hest, Macromol. Biosci., 2008, 8, 991 CrossRef CAS PubMed.
- J. Shi, W. Zhang, X. Wang, Z. Jiang, S. Zhang, X. Zhang, C. Zhang, X. Song and Q. Ai, ACS Appl. Mater. Interfaces, 2013, 5, 5174 CAS.
- X. Wang, J. Shi, Z. Jiang, Z. Li, W. Zhang, X. Song, Q. Ai and H. Wu, Biomacromolecules, 2013, 14, 3861 CrossRef CAS PubMed.
- W. Xu, P. A. Ledin, F. A. Plamper, C. V. Synatschke, A. H. E. Mueller and V. V. Tsukruk, Macromolecules, 2014, 47, 7858 CrossRef CAS.
- C. Ye, Z. A. Combs, R. Calabrese, H. Dai, D. L. Kaplan and V. V. Tsukruk, Small, 2014, 10, 5087 CAS.
- Y. Z. Tian, Y. L. Li, Z. F. Wang and Y. Jiang, J. Mater. Chem. B, 2014, 2, 1667 RSC.
- M. L. Zhu, Y. L. Li, Z. M. Zhang and Y. Jiang, RSC Adv., 2015, 5, 33262 RSC.
- S. N. Pawar and K. J. Edgar, Biomaterials, 2012, 33, 3279 CrossRef CAS PubMed.
- G. Ben Messaoud, L. Sanchez-Gonzalez, A. Jacquot, L. Probst and S. Desobry, J. Colloid Interface Sci., 2015, 440, 1 CrossRef CAS PubMed.
- X. Zeng, W. Wei, X. Li, J. Zeng and L. Wu, Bioelectrochemistry, 2007, 71, 135 CrossRef CAS PubMed.
- B. B. Mandal, S. Kapoor and S. C. Kundu, Biomaterials, 2009, 30, 2826 CrossRef CAS PubMed.
- B. Singh, G. S. Chauhan, S. Kumar and N. Chauhan, Carbohydr. Polym., 2007, 67, 190 CrossRef CAS.
- H. Chen, L. Yuan, W. Song, Z. Wu and D. Li, Prog. Polym. Sci., 2008, 33, 1059 CrossRef CAS.
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133 CrossRef CAS PubMed.
- G. B. Sukhorukov, D. V. Volodkin, A. M. Günther, A. I. Petrov, D. B. Shenoy and H. Möhwald, J. Mater. Chem., 2004, 14, 2073 RSC.
- N. Yamazaki, K. Naganuma, M. Nagai, G. H. Ma and S. Omi, J. Dispersion Sci. Technol., 2003, 24, 249 CrossRef CAS.
- L. W. Chan, H. Y. Lee and P. W. S. Heng, Carbohydr. Polym., 2006, 63, 176 CrossRef CAS.
- J. P. Paques, L. M. C. Sagis, C. J. M. van Rijn and E. van der Linden, Food Hydrocolloids, 2014, 40, 182 CrossRef CAS.
- T. Tanaka and D. J. Fillmore, Journal of Chem. Phys., 1979, 70, 1214 CrossRef CAS.
- T. Tanaka, D. Fillmore, S. T. Sun, I. Nishio, G. Swislow and A. Shah, Phys. Rev. Lett., 1980, 45, 1636 CrossRef CAS.
- J. P. Keener, S. Sircar and A. L. Fogelson, SIAM J. Appl. Math., 2011, 71, 854 CrossRef.
- Y. Hirokawa, E. Sato, S. Hirotsu and T. Tanaka, Abstr. Pap. Am. Chem. S., 1985, 189, 99 Search PubMed.
- C. F. Pratt and D. O. Cooney, AIChE J., 1973, 19, 1049 CrossRef CAS.
- J. L. Mongar and A. Wassermann, Discuss. Faraday Soc., 1949, 118 RSC.
- C. Gelfi and P. G. Righetti, Electrophoresis, 1984, 5, 257 CrossRef CAS.
- V. M. Zasimov, M. G. Golubeva and A. N. Chepurin, Ind. Lab., 1978, 44, 1127 Search PubMed.
- I. Vavruch, Kolloid Z. Z. Polym., 1965, 205, 32 Search PubMed.
- K. I. Draget, B. T. Stokke, Y. Yuguchi, H. Urakawa and K. Kajiwara, Biomacromolecules, 2003, 4, 1661 CrossRef CAS PubMed.
- D. Haznar-Garbacz, G. Garbacz, F. Eisenacher, S. Klein and W. Weitschies, Eur. J. Pharm. Biopharm., 2012, 81, 334 CrossRef CAS PubMed.
- L. Meka, B. Kesavan, V. N. Kalamata, C. M. Eaga, S. Bandari, V. Vobalaboina and M. R. Yamsani, J. Pharm. Sci., 2009, 98, 2122 CrossRef CAS PubMed.
- S. Sungthonjeen, P. Sriamornsak and S. Puttipipatkhachorn, Eur. J. Pharm. Biopharm., 2008, 69, 255 CrossRef PubMed.
- C. G. Zhang, M. Xu, X. G. Tao, J. Y. Tang, Z. T. Liu, Y. Zhang, X. Lin, H. B. He and X. Tang, Int. J. Pharm., 2012, 430, 141 CrossRef CAS PubMed.
- J. Guan, L. Y. Zhou, S. F. Nie, T. X. Yan, X. Tang and W. S. Pan, Int. J. Pharm., 2010, 383, 30 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and additional figures. See DOI: 10.1039/c6ra05711j |
| This journal is © The Royal Society of Chemistry 2016 |