
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
H2 activation using the first 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) :1:1 hetero-tri(aryl)borane†
:1:1 hetero-tri(aryl)borane†
Robin J. Blagg * and
Gregory G. Wildgoose*
* and
Gregory G. Wildgoose*
School of Chemistry, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK. E-mail: r.blagg@uea.ac.uk; g.wildgoose@uea.ac.uk
First published on 28th April 2016
Abstract
The novel 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 hetero-tri(aryl)borane (pentafluorophenyl){3,5-bis(trifluoromethyl)phenyl}(pentachlorophenyl)borane has been synthesised and structurally characterised. This has been show to act as the Lewis acidic component in FLPs for the heterolytic cleavage of H2 with three Lewis bases.
:1:1 hetero-tri(aryl)borane (pentafluorophenyl){3,5-bis(trifluoromethyl)phenyl}(pentachlorophenyl)borane has been synthesised and structurally characterised. This has been show to act as the Lewis acidic component in FLPs for the heterolytic cleavage of H2 with three Lewis bases.
Introduction
Following the initial reports by Stephan and co-workers1,2 there has been rapid growth in the field of frustrated Lewis pair (FLP) chemistry.3–9 With the archetypal system of the electron-poor sterically bulky Lewis acid tris(pentafluorophenyl)borane, B(C6F5)3 and the electron-rich bulky Lewis base tri(tert-butyl)phosphine, P(tBu)3, able to effect the heterolytic cleavage of H2 under mild conditions; generating both hydridic and protic products.The activation of H2 by FLPs has led to them being used as mediators or catalysts for metal-free hydrogenation for a range of functional groups, examples include; alkenes and alkynes,10,11 aldehydes and ketones,12–14 and imines.15 Although these and other FLP systems have used a wide range of Lewis bases as stoichiometric reagents, substrates, and solvents, the vast majority continue to use B(C6F5)3 or related halogenated tri(aryl)boranes as the Lewis acidic component. However, the tri(aryl)boranes are not the only Lewis acids which have been used in FLPs. Other examples include N-heterocycle carbene stabilised borenium cations,16,17 aluminium analogues of the tri(aryl)boranes,18 carbon-based N-methylacridinium cations,19,20 and phosphorus(III)-based N-heterocyclic phosphenium cations.21
A number of systematic studies on tri(aryl)boranes have been reported that incorporate the stepwise substitution of aryl groups in the archetypal borane, B(C6F5)3. For example, the series B(2,4,6-Me3C6H2)n(C6F5)3−n (n = 1–3),22 and B(C6Cl5)n(C6F5)3−n (n = 1–3)23 were used in both cases to predict the reduction potential of B(C6F5)3, which was subsequently reported from direct measurements by ourselves.24,25 Although three-coordinate boranes with three different substituents are known, such as the bis(aryl)boryl ferrocene FcB(C6Cl5)(2,4,6-Me3C6H2);26 there are as yet no examples where all three substituents are phenyl rings.
In an effort to expand the range of Lewis acidic boranes for FLP reactions, we have previously reported studies of homo-tri(aryl)boranes {aryl = C6F5, C6Cl5, 3,5-(CF3)2C6H3, 2,4-(CF3)2C6H3, 2,5-(CF3)2C6H3}24,25,27,28 and 2:1 hetero-tri(aryl)boranes {aryl = C6F5/ArF5, C6Cl5/ArCl5, 3,5-(CF3)2C6H3/ArF6}.29 In these publications we also introduced the concept of using the standard reduction potential of tri(aryl)boranes as a proxy measure of their Lewis acidities; such a method is base independent, and measures a property of the trigonal-planar reactive species.
Herein we report the synthesis and characterisation of (pentafluorophenyl){3,5-bis(trifluoromethyl)phenyl}(pentachlorophenyl)borane 3, completing the B(ArF5)x(ArF6)y(ArCl5)z family, which to the best of our knowledge is the first structurally characterised 1:1:1 hetero-tri(aryl)borane.30 This not only demonstrate the applicability of the synthetic methodology used; but is then studied as the Lewis acidic component in a series of FLPs for H2 activation under mild conditions, and together with the associated tri(aryl)borohydride studied electrochemically.
Results and discussion
In order to synthesise B(C6F5){3,5-(CF3)2C6H3}(C6Cl5) 3 (Scheme 1), we initially adapted the stepwise route for the synthesis of B{3,5-(CF3)2C6H3}2Br via B{3,5-(CF3)2C6H3}2(OMe) of Samigullin et al.,31 for the stepwise synthesis of B(C6F5){3,5-(CF3)2C6H3}(OMe) 1. This entailed the generation of Li(C6F5) at −78 °C, and its reaction in Et2O with BH3·SMe2 to give [Li(OEt2)n][H3B(C6F5)], followed by hydride abstraction using one equivalent Me3SiCl to give BH2(C6F5). The bis(trifluoromethyl)phenyl ring was then added by reaction with Li{3,5-(CF3)2C6H3} at −78 °C to give [Li(OEt2)n][H2B(C6F5){3,5-(CF3)2C6H3}], with hydride-abstraction by Me3SiCl giving BH(C6F5){3,5-(CF3)2C6H3}. Reaction with excess methanol then converts the hydride to the methoxide 1 (while the aryl rings may be added in either order, less by-products were observed when the C6F5 ring was added first). Conversion of the methoxide to the bis(aryl)bromoborane was readily achieved by reaction of 1 with excess BBr3, leading to B(C6F5){3,5-(CF3)2C6H3}Br 2 being obtained as a pale yellow oil.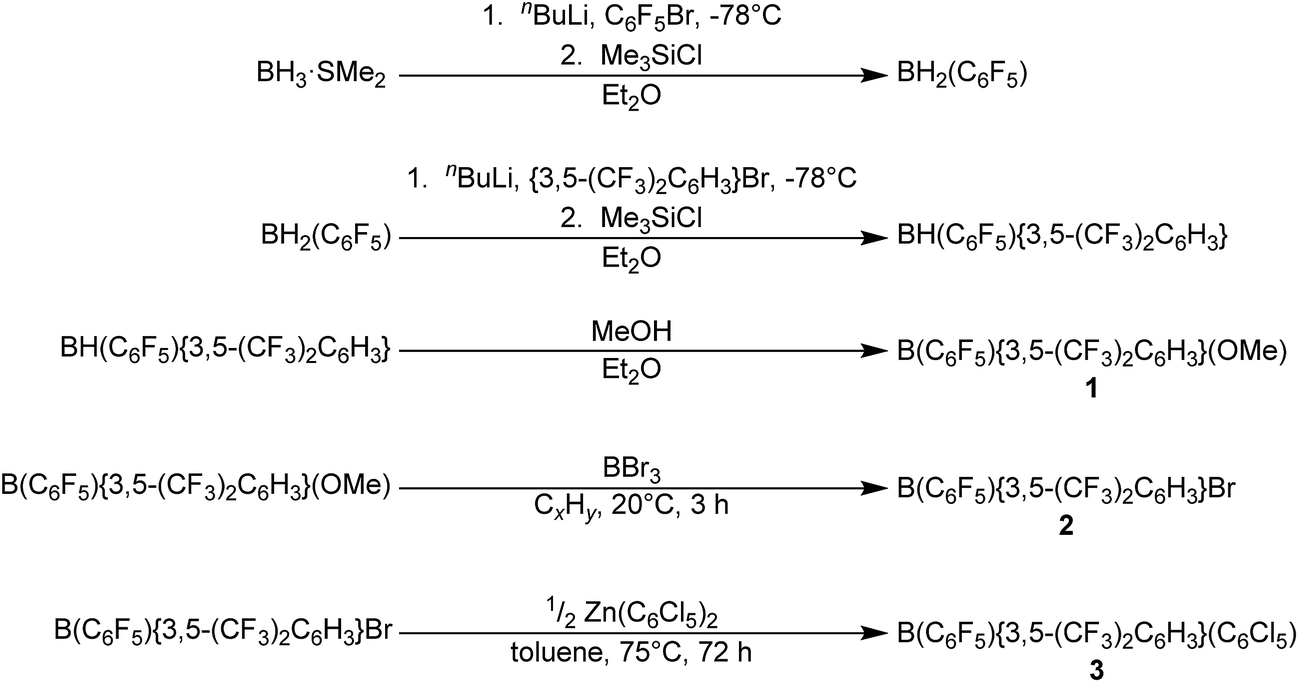 | ||
| Scheme 1 Synthetic route for the synthesis of B(C6F5){3,5-(CF3)2C6H3}(C6Cl5) 3. | ||
Reacting 2 with half an equivalent of Zn(C6Cl5)2 in hot toluene, and isolation of the hydrocarbon soluble product leads to isolation of crude 3 as a yellow-brown oil (from which yellow crystals of the product will eventually form). The oil may be purified by removal of impurities via sublimation; followed by dissolving the oil in a minimum volume of CH2Cl2, addition of n-hexane, concentration in vacuo to a minimum volume and cooling to −25 °C. This allows for the isolation of 3 as a microcrystalline yellow solid.
The X-ray crystal structure of 3 was obtained from single crystals obtained from the slow crystallisation of the crude oil at room temperature (Fig. 1 & S1,† Table 1). The structure obtained comprises two crystallographically unique examples of 3 in the asymmetric unit, with negligible differences in structural metrics.
 | ||
| Fig. 1 X-ray crystallographic structure of B(C6F5){3,5-(CF3)2C6H3}(C6Cl5) 3. | ||
| B(C6F5){3,5-(CF3)2C6H3}(C6Cl5) 3 | |
|---|---|
| Empirical formula | C20H3B1Cl5F11 |
| Formula weight | 640.28 |
| Temperature/K | 140(3) |
| Crystal system | Triclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/Å | 8.9724(11) |
| b/Å | 15.5705(16) |
| c/Å | 17.2632(17) |
| α/° | 86.822(8) |
| β/° | 86.077(9) |
| γ/° | 74.230(10) |
| Volume/Å3 | 2313.8(4) |
| Z | 4 |
| ρcalc/mg mm−3 | 1.838 |
| μ/mm−1 | 0.725 |
| F(000) | 1248.0 |
| Crystal size/mm3 | 0.1 × 0.05 × 0.05 |
| Radiation | Mo Kα (λ = 0.7107 Å) |
| 2θ range for data collection | 6.44 to 52.74° |
| Index ranges | −11 ≤ h ≤ 7, −19 ≤ k ≤ 18, −21 ≤ l ≤ 21 |
| Reflections collected | 14047 |
| Independent reflections | 9352 [Rint = 0.0907, Rsigma = 0.3709] |
| Data/restraints/parameters | 9352/0/667 |
| Goodness-of-fit on F2 | 0.950 |
| Final R indexes [I ≥ 2σ(I)] | R1 = 0.0982, wR2 = 0.0919 |
| Final R indexes [all data] | R1 = 0.3359, wR2 = 0.1515 |
| Largest diff. peak/hole/e A−3 | 0.51/−0.37 |
The structure of 3, is consistent with those previously reported for tri(aryl)boranes;23,29 a trigonal-planar BC3 centre with the aryl rings twisted about their B–C bonds to minimise steric interactions. The degree of twist increasing with the size of the ortho-substituent on the aryl ring: 21(3), 43(1), 83(2)° {for the ArF6 (o-H), ArF5 (o-F), ArCl5 (o-Cl) rings respectively}. As this angle increases there are two corresponding effects, affecting the Lewis acidity of the boron centre. Firstly, it reduces the overlap between the filled π orbitals of the aryl ring with the formally vacant boron 2pz orbital, suggesting small angles would lead to attenuation of Lewis acidity. While increased twist angles will increases the sterics around the boron centre and the chance of donation of electron density from lone pairs on the ortho-substituents into the boron 2pz orbital (the significant twist of the ArCl5 ring in 3, coupled with the size of the chlorine atoms show potential for such donation, B…o-Cl ca. 3.0 Å). This attenuation of the Lewis acidity of the borane; has been previously identified electrochemically for boranes incorporating ortho-CF3 and ortho-Cl substituents on the phenyl rings.25,29
Cyclic voltammograms were obtained at varying scan rates for 3 (Fig. 2), in the weakly-coordinating solvent/electrolyte CH2Cl2/[nBu4N][B(C6F5)4] at a glassy carbon electrode. Digital simulations of the experimental voltammetric data modelled using an EC-mechanism (i.e. a reversible, heterogeneous electron transfer step followed by an irreversible, homogeneous chemical step which generates electro-inactive products), allow us to extract pertinent mechanistic parameters. Specifically, the formal redox potential and charge transfer coefficient (E° and α respectively, where E° is a measure of the electrophilicity and hence a proxy for measurement of Lewis acidity at the boron centre),29 and kinetic parameters for the electron transfer (k0) and follow-on chemical step (kf).
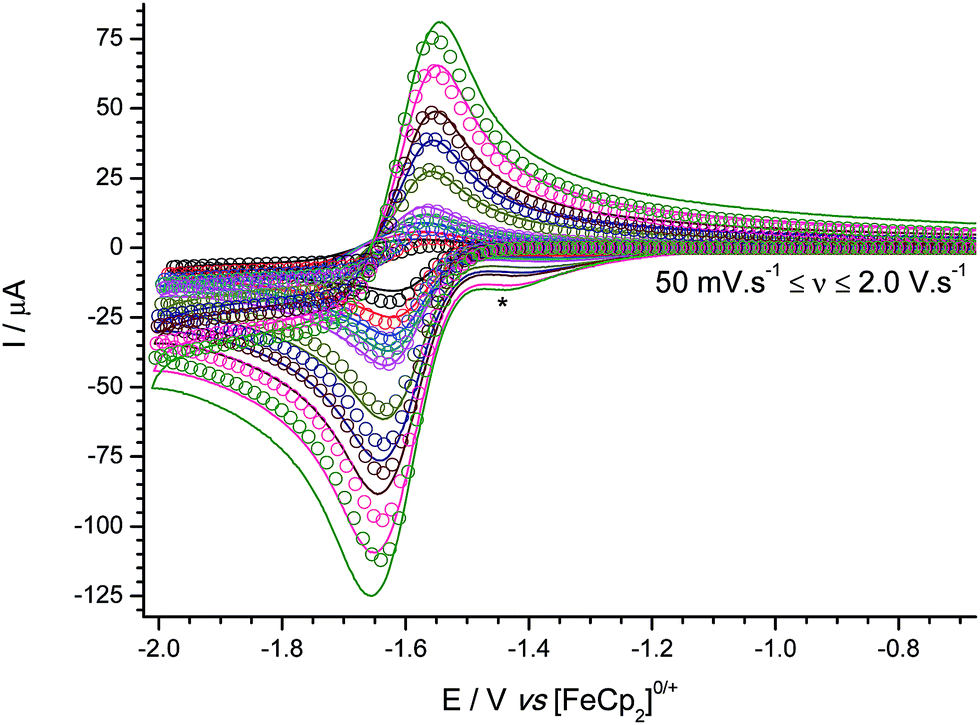 | ||
| Fig. 2 Experimental (line) and simulated (open circles) cyclic voltammograms for the reduction of B(C6F5){3,5-(CF3)2C6H3}(C6Cl5) 3. Simulated using an EC-mechanism with: E° = −1.60 ± 0.01 V vs. [FeCp2]0/+; α = 0.499; k0 = 4.35 × 10−2 cm s−1; kf = 0.47 s−1 (modelled as a pseudo first-order process); D(30/˙−) = 1.10 × 10−5 cm2 s−1, D obtained via 1H and 19F DOSY NMR spectroscopy. * Trace impurity in solvent/electrolyte. | ||
In comparison with our previously reported electrochemical data for homo- and 2:1 hetero-tri(aryl)boranes,25,29 the formal redox potential is comparable to those of B{3,5-(CF3)2C6H3}3, and B(C6Cl5)2{3,5-(CF3)2C6H3} (−1.61 and −1.60 V, respectively); more negative than those of the B(C6F5)x(C6Cl5)z, and remaining B(C6F5)x{3,5-(CF3)2C6H3}y series (−1.58 to −1.52 V); with only B{3,5-(CF3)2C6H3}2(C6Cl5), B{2,4-(CF3)2C6H3}3, and B{2,5-(CF3)2C6H3}3 exhibiting more negative redox potentials (−1.70, −1.79 and −1.85 V, respectively). As with all the boranes incorporating at least one pentachlorophenyl ring, the associated radical anion is relatively stable (as measured by the rate, kf, of the decomposition of the radical anion intermediate), due to the bulky ring(s), preventing the boron-centred radical from being accessible for further reaction (assumed to be with the solvent).
For the sake of completeness, the Lewis acidity was also quantified using the common “Gutmann–Beckett” method,32,33 where the 31P and 11B NMR shifts for the adduct Et3PO–3 in CD2Cl2 are +76.49 and +2.51 ppm respectively. Corresponding to a “Gutmann–Beckett acceptor number” of 78.4 (calculated as detailed in ref. 34); significantly different to that of B{3,5-(CF3)2C6H3}3, 83.8, which exhibits a comparable formal redox potential.25,35
The potential for 3 to act as the Lewis acidic component of an FLP for the cleavage of H2 in dichloromethane at ca. +20 °C was screened for, together with the phosphorus- and nitrogen-based Lewis bases, P(tBu)3, 2,2,6,6-tetramethylpiperidine (tmp) and 2,6-lutidine; all of which have been shown to cleave H2 in conjunction with B(C6F5)3.2,36,37 The reactions were monitored by 1H and 11B NMR spectroscopy (see Fig. S2–4†). In all three cases irreversible H2 cleavage38 occurred leading to the formation of the terminal tri(aryl)borohydride [H–3]−. The conversions to [H–3]− were slow and relatively clean (<10% by-products by 1H & 11B NMR spectroscopy); allowing for the percentage conversion to be monitored by 1H NMR spectroscopy (Fig. S5†). This showed the FLPs 3/P(tBu)3 and 3/lutidine underwent comparable percentage conversions with time (ca. 60% conversion after 7 days), while the 3/tmp system proceeded slower (38% conversion after 7 days).
Two other features of interest were noted as the reactions progressed. Firstly, in the 3/tmp and 3/lutidine systems distinct resonances corresponding to the free and protonated bases were not observed {while they were for the 3/P(tBu)3 system}, therefore suggesting rapid equilibrium between free and protonated bases in solution. Secondly, while resonances associated with [H–3]− generated from the 3/P(tBu)3 and 3/lutidine FLPs matched (δH +4.08, δB −14.3, 1JHB 88 Hz), those from the 3/tmp FLP displayed slight shifts (δH +3.98, δB −13.9, 1JHB 84 Hz). Such differences are comparable to those observed between [(tBu)3PH][HB(C6F5)2(C6Cl5)] (δH +3.94, δB −19.6, 1JHB 90 Hz)29 and [tmp–H][HB(C6F5)2(C6Cl5)] (δH +3.81, δB −19.1, 1JHB 84 Hz),39 a species which has been established (in the solid state) as retaining a close contact between the hydridic and protic hydrogens.40 While this is not enough to claim that [tmp–H][HB(C6F5)2(C6Cl5)] and [tmp–H][H–3] retain such interaction in solution; the [tmp–H]+ cation must be interacting differently from the others with the anion for these changes in the spectral data of the tri(aryl)borohydrides to be observed.
The tri(aryl)borohydride anion can also be directly synthesised as the tetrabutylammonium salt, by reaction of 3 with Na[HBEt3] in toluene in the presence of [nBu4N]Cl, followed by isolation from Et2O/n-hexane, to give [nBu4N][H–3] as a pale yellow oil (the non-symmetric anion [H–3]− leading to formation of a room temperature ionic liquid). NMR spectral data for [nBu4N][H–3] matches those of [(tBu)3PH][H–3] and [lutidine–H][H–3] obtained via the FLP reactions with H2.
Cyclic voltammograms obtained of [nBu4N][H–3] (Fig. 3, under the same conditions used for 3) show a single irreversible oxidation wave, with a peak potential (at 100 mV s−1) of +1.07 V vs. [FeCp2]0/+; with no clear observation of subsequent regeneration of the parent borane 3. These voltammograms can be adequately simulated using a comparable ECE-mechanism to that previously used for [HB(C6F5)3]−.27 Specifically, a one-electron oxidation of [H–3]− to the neutral radical [H–3]˙, followed by rapid loss of H+ to generate the radical-anion 3˙−, which due to the strongly oxidising potential is rapidly converted to 3. With the liberated H+ reacting with the regenerated 3, leading to its decomposition to electrochemically inactive species, hence it not being observed electrochemically.
 | ||
| Fig. 3 Experimental (line) and simulated (open circles) cyclic voltammograms for the oxidation of [HB(C6F5){3,5-(CF3)2C6H3}(C6Cl5)]− [H–3]− as a [nBu4N]+ salt. Simulated using an ECE-mechanism with: E°(1) = +0.93 ± 0.01 V vs. [FeCp2]0/+; α(1) = 0.500; k0(1) = 7.60 × 10−4 cm s−1; kf(2) = 6.38 × 103 s−1 (modelled as a pseudo first-order process); E°(3) = −1.60 ± 0.01 V vs. [FeCp2]0/+; α(3) = 0.501; k0(3) = 4.35 × 10−2 cm s−1; D([H–3]−/˙) = 1.06 × 10−5 cm2 s−1, D obtained via 1H DOSY NMR spectroscopy. | ||
Compared to our previously reported electrochemical data for tri(aryl)borohydrides, all of which also give a single irreversible oxidation wave; the peak potential for [H–3]− is higher than that of [HB(C6F5)3]− (+0.88 V),27 comparable to that of [HB{3,5-(CF3)2C6H3}3]− (+1.08 V, as a Na+ salt), and lower than those of [HB{2,4-(CF3)2C6H3}3]− and [HB{2,5-(CF3)2C6H3}3]− (+1.31 and +1.13 V, respectively, as Na+ salts) 25 (all potentials verses [FeCp2]0/+ at 100 mV s−1). As with all these boranes this shows an improvement over the direct oxidation of H2 (Ep > +1.5 V vs. [FeCp2]0/+ at 100 mV s−1) under these conditions.27
Conclusions
The synthesis of the first structurally characterised 1:1:1 hetero-tri(aryl)borane, by a stepwise methodology, is but the first example whereby this or similar methodologies can be used to greatly expand the accessible range of Lewis acids. Thereby giving potential for fine tuning of the properties of such species and hence control of resulting reactivities.
Demonstrating FLP hydrogen cleavage with three Lewis bases, showed that the percentage conversion to cleaved products over time is dependent on the choice of Lewis base for a specific Lewis acid. Further while [(tBu)3PH]+, [lutidine–H]+, and [nBu4N]+, all appear to act as non-interacting cations with respect of the tri(aryl)borohydride; subtle differences observed by NMR spectroscopy suggests this is not the case for [tmp–H]+, such a difference could be reflected in the reactivities of these hydrogen cleavage products.
Electrochemical studies allowed for not only the direct measurement of the electrophilicity of the free borane, but also quantification as to the stability of the associated radical-anion, and measurements of the associated borohydride assessing potential for regeneration of the parent borane following net two electron oxidation and proton liberation.
Experimental
All reactions and manipulations were performed under an atmosphere of dry oxygen-free N2, using either standard Schlenk techniques or in an MBraun UNIlab glovebox. All solvents were dried prior to use by refluxing over an appropriate drying agent {Na/benzophenone for petroleum ether (bp 40–60 °C), diethyl ether, n-hexane; Na for toluene; CaH2 for dichloromethane}, collected by distillation under a dry oxygen-free N2 atmosphere and stored over 4 Å molecular sieves prior to use.NMR spectra were obtained on a Bruker Avance DPX-500 spectrometer; for 1H spectra residual protio-solvent was used as an internal standard; for 13C the solvent resonance was used as an internal standard;41 for 19F spectra CFCl3 was used as an external standard; for 11B spectra BF3·OEt2 was used as an external standard; for 31P spectra 85% H3PO4 was used as an external standard.
Mass spectrometry was performed by the EPSRC Mass Spectrometry Service at the University of Swansea. Elemental analyses were performed by the Elemental Analysis Service at London Metropolitan University.
Single crystals of 3 were grown by slow crystallisation of the crude oil at room temperature; suitable crystals were selected, encapsulated in a viscous perfluoropolyether and mounted on an Agilent Technologies Xcaliber-3 single crystal X-ray diffractometer using Mo Kα radiation, where the crystals were cooled to 140 K during data collection and a full sphere of data collected. The data was reduced and an absorption correction performed using Agilent Technologies CrysAlisPro.42 Using Olex2,43 the structure was solved and space group assigned with SuperFlip/EDMA using charge flipping,44 and then refined with the ShelXL version 2014/7 refinement program using least squares minimisation.45
Due to poor crystal quality and resulting weak diffraction, the crystallographic statistics for 3 are less than ideal. This does not affect any conclusions drawn herein based on this data.
CCDC 1446563 contains the supplementary crystallographic data for this paper.†
Electrochemical studies were carried out using a Metrohm Autolab PGSTAT302N potentiostat linked to a computer running Metrohm Autolab NOVA version 1.11 software, in conjunction with a three electrode cell. The working electrode was a glassy carbon disc (Bioanalytical Systems, Inc., ca. 7.0 mm2 area calibrated using the [FeCp2]0/+ redox couple); the counter electrode was a platinum wire (99.99% purity); and the pseudo-reference electrode a silver wire (99.99% purity). All electrodes were polished with 0.3 μm α-alumina slurry, washed, and dried prior to use. All electrochemical measurements were performed at ambient temperature under a dry N2 atmosphere, in CH2Cl2 containing 50 μmol cm−3 [nBu4N][B(C6F5)4] as the supporting electrolyte and 1.0 to 2.0 μmol cm−3 of the analyte species of interest, and iR-compensated using positive-feedback to within 85 ± 5% of the uncompensated solution resistance. [nBu4N][B(C6F5)4] was synthesised according to published methods.46 All potentials were referenced to the [FeCp2]0/+ redox couple, which was added as an internal standard. Simulations of electrochemical processes were performed using ElchSoft DigiElch – Professional version 7.FD software.47
Zn(C6Cl5)2 was synthesised as previously reported.23 All other reagents were obtained from commercial suppliers and used as supplied.
B(C6F5){3,5-(CF3)2C6H3}(OMe) 1
nBuLi (15.6 cm3, 25 mmol, 1.6 mmol cm−3 in hexanes) was added to a cooled (−78 °C) solution of bromopentafluorobenzene (8.02 cm3, 25 mmol) in Et2O (50 cm3) and stirred for 10 min. BH3·SMe2 (2.37 cm3, 25 mmol) is added, and after 10 min the reaction mixture warmed to room temperature and stirred for a further 30 min. Me3SiCl (3.17 cm3, 25 mmol) is added to the clear pale yellow solution, resulting in the rapid formation of a white precipitate, the mixture is stirred for 30 min then cooled to −78 °C. 3,5-Bis(trifluoromethyl)bromobenzene (5.80 cm3, 25 mmol) is added, followed by nBuLi (15.6 cm3, 25 mmol, 1.6 mmol cm−3 in hexanes), after 10 min the reaction mixture warmed to room temperature and stirred for a further 30 min. Me3SiCl (3.17 cm3, 25 mmol) is added, and the mixture is stirred for 30 min. Methanol (3.0 cm3, 74.2 mmol) is slowly added, resulting in rapid evolution of H2, and the mixture stirred for an hour. Volatiles are removed in vacuo, and the product extracted into petroleum ether (50 cm3), isolated by filtration (via cannula) as a clear pale yellow solution. Removal of volatiles in vacuo gives the product as an off white solid. Yield 6.10 g (14.5 mmol, 58%). 1H NMR (500.2 MHz, C6D6, 25 °C, δ): +8.14 (s, 2H, ArF6 2,6-H), +7.88 (s, 1H, ArF6 4-H), +3.30 (s, 3H, OMe); 11B NMR (160.5 MHz, C6D6, 25 °C, δ): +42.1 (br.s); 13C{1H} NMR (125.8 MHz, C6D6, 25 °C, δ): +146.8 (br.d, 1JCF ≈ 244 Hz, ArF5 2,6-C), +143.1 (br.d, 1JCF ≈ 256 Hz, ArF5 4-C), +138.2 (br.d, 1JCF ≈ 254 Hz, ArF5 3,5-C), +135.3 (br.q, 3JCF = 3.7 Hz, ArF6 2,6-C), +132.1 (q, 2JCF = 33.0 Hz, ArF6 3,5-C), +126.5 (sept., 3JCF = 3.7 Hz, ArF6 4-C), +124.3 (q, 1JCF = 273 Hz, ArF6 3,5-CF3), +57.0 (s, OMe); 19F NMR (470.7 MHz, C6D6, 25 °C, δ): −63.0 (s, 6F, ArF6 3,5-CF3), −131.6 (m, 2F, ArF5 2,6-F), −149.1 (t, 3JFF = 21.2 Hz, 1F, ArF5 4-F), −159.7 (m, 2F, ArF5 3,5-F). 1H NMR (500.2 MHz, CD2Cl2, 25 °C, δ): +8.16 (s, 2H, ArF6 2,6-H), +8.04 (s, 1H, ArF6 4-H), +4.00 (s, 3H, OMe); 11B NMR (160.5 MHz, CD2Cl2, 25 °C, δ): +42.4 (br.s); 19F NMR (470.7 MHz, CD2Cl2, 25 °C, δ): −63.3 (s, 6F, ArF6 3,5-CF3), −131.3 (m, 2F, ArF5 2,6-F), −150.8 (t, 3JFF = 19.9 Hz, 1F, ArF5 4-F), −160.5 (m, 2F, ArF5 3,5-F).Warning: LiC6F5 is known to be thermally unstable (explosive above ca. −20 °C). It is essential to maintain the reaction mixture at −78 °C until the reaction with BH3·SMe2 is completed.
B(C6F5){3,5-(CF3)2C6H3}Br 2
To a colourless solution of 1 (1.56 g, 3.70 mmol) in petroleum ether (10 cm3) is added BBr3 (ca. 0.4 cm3, 4.2 mmol) and the mixture stirred for 3 hours. All volatiles are removed in vacuo from the cloudy pale yellow solution, to give the product as a cloudy yellow oil. 1H NMR (500.2 MHz, CD2Cl2, 25 °C, δ): +8.44 (s, 2H, ArF6 2,6-H), +8.23 (s, 1H, ArF6 4-H); 11B NMR (160.5 MHz, CD2Cl2, 25 °C, δ): +64.5 (br.s); 13C{1H} NMR (125.8 MHz, CD2Cl2, 25 °C, δ): +146.6 (br.d, 1JCF ≈ 250 Hz, ArF5 2,6-C), +138.5 (br.d, 1JCF ≈ 256 Hz, ArF5 3,5-C), +137.8 (br.q, 3JCF = 3.8 Hz, ArF6 2,6-C), +132.4 (q, 2JCF = 33.9 Hz, ArF6 3,5-C), +129.3 (sept., 3JCF = 3.8 Hz, ArF6 4-C), +123.6 (q, 1JCF = 273 Hz, ArF6 3,5-CF3); 19F NMR (470.7 MHz, CD2Cl2, 25 °C, δ): −63.3 (s, 6F, ArF6 3,5-CF3), −128.8 (m, 2F, ArF5 2,6-F), −147.8 (tt, 3JFF = 19.9 Hz, 4JFF = 3.6 Hz, 1F, ArF5 4-F), −160.5 (m, 2F, ArF5 3,5-F).B(C6F5){3,5-(CF3)2C6H3}(C6Cl5) 3
To a colourless solution of 1 (2.08 g, 4.93 mmol) in petroleum ether (15 cm3) is added BBr3 (ca. 0.5 cm3, 5.2 mmol) and the mixture stirred for 3 hours. All volatiles are removed in vacuo from the cloudy pale yellow solution giving a cloudy yellow oil, 2. Zn(C6Cl5)2 (1.39 g, 2.46 mmol) is added and the mixture dissolved/suspended in toluene (10 cm3). The mixture is stirred at +75 °C for ca. 66 hours to give a cloudy yellow solution. Volatiles are removed in vacuo, and the crude product extracted into petroleum ether and isolated by filtration (via cannula) as a clear golden yellow solution, volatiles are removed in vacuo to give a cloudy orange oil. Impurities may be removed by sublimation at 90 °C/10−1 mbar, and the oil recrystallized from CH2Cl2/n-hexane at −25 °C as a yellow micro-crystalline solid. Yield 0.77 g (1.20 mmol, 58%). 1H NMR (500.2 MHz, CD2Cl2, 25 °C, δ): +8.20 (s, 1H, ArF6 4-H), +8.14 (s, 2H, ArF6 2,6-H); 11B NMR (160.5 MHz, CD2Cl2, 25 °C, δ): +63.4 (br.s); 13C{1H} NMR (125.8 MHz, CD2Cl2, 25 °C, δ): +149.1 (br.d, 1JCF ≈ 252 Hz, ArF5 2,6-C), +145.5 (br.d, 1JCF ≈ 261 Hz, ArF5 4-C), +140.7 (br.m, ArF6/Cl5 1-C), +139.8 (br.m, ArF6/Cl5 1-C), +138.5 (br.d, 1JCF ≈ 253 Hz, ArF5 3,5-C), +137.8 (br.q, 3JCF = 3.2 Hz, ArF6 2,6-C) +135.7 (s, ArCl5 4-C), +132.9 (s, ArCl5 2,6/3,5-C), +132.5 (q, 2JCF = 33.4 Hz, ArF6 3,5-C), +132.2 (s, ArCl5 2,6/3,5-C), +129.0 (sept., 3JCF = 3.8 Hz, ArF6 4-C), +123.7 (q, 1JCF = 273 Hz, ArF6 3,5-CF3), +111.7 (br.m, ArF5 1-C); 19F NMR (470.7 MHz, CD2Cl2, 25 °C, δ): −63.3 (s, 6F, ArF6 3,5-CF3), −125.8 (br.m, 2F, ArF5 2,6-F), −144.7 (t, 3JFF = 19.9 Hz, 1F, ArF5 4-F), −160.6 (m, 2F, ArF5 3,5-F). HRMS-APCI (m/z): [M]+ calc. for C20H3BCl5F11, 639.8565; found, 639.8565. Elemental analysis (calc. for C20H3BCl5F11): C 37.66 (37.52), H 0.44 (0.47).[nBu4N][HB(C6F5){3,5-(CF3)2C6H3}(C6Cl5)] [nBu4N][H–3]
To a solution of 3 (250 mg, 0.39 mmol) and [nBu4N]Cl (109 mg, 0.39 mmol) in toluene (10 cm3) is added Na[HBEt3] (0.4 cm3, 0.4 mmol, 1.0 mmol cm−3 in toluene), the solution stirred for two hours, then all volatiles removed in vacuo. The product is extracted into Et2O, filtered, and isolated by addition n-hexane, concentration in vacuo, and cooling at −25 °C. To give a pale yellow oil. Yield 273 mg (0.31 mmol, 79%). 1H NMR (500.2 MHz, CD2Cl2, 25 °C, δ): +7.69 (s, 2H, ArF6 2,6-H), +7.50 (s, 1H, ArF6 4-H), +4.12 (br.q, 1JHB = 86 Hz, 1H), +3.00 (m, 8H, nBu), +1.55 (m, 8H, nBu), +1.33 (m, 8H, nBu), +0.95 (t, 3JHH = 7.4 Hz, 12H, nBu); 11B NMR (160.5 MHz, CD2Cl2, 25 °C, δ): −14.2 (d, 1JBH = 86 Hz); 13C{1H} NMR (125.8 MHz, CD2Cl2, 25 °C, δ): +160.5 (br.m, Ar? 1-C), +156.2 (br.m, Ar? 1-C), +148.8 (dm, 1JCF ≈ 235 Hz, ArF5 2,6-C), +139.0 (s, ArCl5), +138.3 (dm, 1JCF ≈ 244 Hz, ArF5 4-C), +137.3 (dm, 1JCF ≈ 247 Hz, ArF5 3,5-C), +134.3 (br.q, 3JCF = 3.3 Hz, ArF6 2,6-C), +130.9 (s, ArCl5), +129.3 (s, ArCl5), +128.8 (q, 2JCF = 30.7 Hz, ArF6 3,5-C), +127.2 (br.m, Ar? 1-C), +124.4 (q, 1JCF = 272 Hz, ArF6 3,5-CF3), +117.2 (sept., 3JCF = 4.1 Hz, ArF6 4-C), +59.5 (t, 1JCN = 2.7 Hz, nBu CH2), +24.3 (s, nBu CH2), +20.1 (s, nBu CH2), +13.7 (s, nBu CH3); 19F NMR (470.7 MHz, CD2Cl2, 25 °C, δ): −62.3 (s, 6F, ArF6 3,5-CF3), −130.9 (br.m, 2F, ArF5 2,6-F), −164.4 (t, 3JFF = 20.3 Hz, 1F, ArF5 4-F), −167.0 (m, 2F, ArF5 3,5-F). Elemental analysis (calc. for C36H40BCl5F11N): C 48.63 (48.92), H 4.45 (4.56), N 1.47 (1.58).Acknowledgements
G. G. W. thanks the Royal Society for financial support by a University Research Fellowship. The research leading to these results has received funding from the European Research Council under the ERC Grant Agreement no. 307061 (PiHOMER). We acknowledge the use of the EPSRC funded National Chemical Database Service hosted by the Royal Society of Chemistry, and the EPSRC UK National Mass Spectrometry Facility (NMSF) at the University of Swansea.References
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef CAS PubMed.
- D. W. Stephan, Org. Biomol. Chem., 2008, 6, 1535–1539 CAS.
- D. W. Stephan, Dalton Trans., 2009, 3129–3136 RSC.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- D. W. Stephan, Compr. Inorg. Chem. II, 2013, 1, 1069–1103 CAS.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed.
- L. J. Hounjet, C. Bannwarth, C. N. Garon, C. B. Caputo, S. Grimme and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 7492–7495 CrossRef CAS PubMed.
- M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 8396–8397 CrossRef CAS PubMed.
- T. Mahdi and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 15809–15812 CrossRef CAS PubMed.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, J. Am. Chem. Soc., 2014, 136, 15813–15816 CrossRef CAS PubMed.
- Á. Gyömöre, M. Bakos, T. Földes, I. Pápai, A. Domján and T. Soós, ACS Catal., 2015, 5, 5366–5372 CrossRef.
- P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 8050–8053 CrossRef CAS PubMed.
- J. M. Farrell, J. A. Hatnean and D. W. Stephan, J. Am. Chem. Soc., 2012, 134, 15728–15731 CrossRef CAS PubMed.
- E. J. Lawrence, T. J. Herrington, A. E. Ashley and G. G. Wildgoose, Angew. Chem., Int. Ed., 2014, 53, 9922–9925 CrossRef CAS PubMed.
- G. Ménard and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 53, 8272–8275 CrossRef PubMed.
- E. R. Clark and M. J. Ingleson, Angew. Chem., Int. Ed., 2014, 53, 11306–11309 CrossRef CAS PubMed.
- E. J. Lawrence, E. R. Clark, L. D. Curless, J. M. Courtney, R. J. Blagg, M. J. Ingleson and G. G. Wildgoose, Chem. Sci., 2016, 7, 2537–2543 RSC.
- J. M. Bayne and D. W. Stephan, Chem. Soc. Rev., 2016, 45, 765–774 RSC.
- S. A. Cummings, M. Iimura, C. J. Harlan, R. J. Kwaan, I. V. Trieu, J. R. Norton, B. M. Bridgewater, F. Jäkle, A. Sundararaman and M. Tilset, Organometallics, 2006, 25, 1565–1568 CrossRef CAS.
- A. E. Ashley, T. J. Herrington, G. G. Wildgoose, H. Zaher, A. L. Thompson, N. H. Rees, T. Krämer and D. O'Hare, J. Am. Chem. Soc., 2011, 133, 14727–14740 CrossRef CAS PubMed.
- E. J. Lawrence, V. S. Oganesyan, G. G. Wildgoose and A. E. Ashley, Dalton Trans., 2013, 42, 782–789 RSC.
- R. J. Blagg, E. J. Lawrence, K. Resner, V. S. Oganesyan, T. J. Herrington, A. E. Ashley and G. G. Wildgoose, Dalton Trans., 2016, 45, 6023–6031 RSC.
- M. J. Kelly, R. Tirfoin, J. Gilbert and S. Aldridge, J. Organomet. Chem., 2014, 769, 11–16 CrossRef CAS.
- E. J. Lawrence, V. S. Oganesyan, D. L. Hughes, A. E. Ashley and G. G. Wildgoose, J. Am. Chem. Soc., 2014, 136, 6031–6036 CrossRef CAS PubMed.
- E. J. Lawrence, R. J. Blagg, D. L. Hughes, A. E. Ashley and G. G. Wildgoose, Chem.–Eur. J., 2015, 21, 900–906 CrossRef PubMed.
- R. J. Blagg, T. R. Simmons, G. R. Hatton, J. M. Courtney, E. L. Bennett, E. J. Lawrence and G. G. Wildgoose, Dalton Trans., 2016, 45, 6032–6043 RSC.
- The Cambridge structural database (CSD version 5.37) was searched for all structures containing the fragment B(C6)3, while not including the fragments: B(C6H5)2, B(C6F5)2, B{3,5-(CF3)2C6H3}2, B{2,4,6-Me3C6H2}2, or B(C6)3 (any atom); the 152 structures such identified were then inspected manually, confirming no 1:1:1 hetero-tri(aryl)boranes present.
- K. Samigullin, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2014, 33, 3564–3569 CrossRef CAS.
- U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235–1257 CrossRef CAS.
- M. A. Beckett, G. C. Strickland, J. R. Holland and K. S. Varma, Polymer, 1996, 37, 4629–4631 CrossRef CAS.
- I. B. Sivaev and V. I. Bregadze, Coord. Chem. Rev., 2014, 270–271, 75–88 CrossRef CAS.
- T. J. Herrington, A. J. W. Thom, A. J. P. White and A. E. Ashley, Dalton Trans., 2012, 41, 9019–9022 RSC.
- V. Sumerin, F. Schulz, M. Nieger, M. Leskelä, T. Repo and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 6001–6003 CrossRef CAS PubMed.
- S. J. Geier and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 3476–4347 CrossRef CAS PubMed.
- The irreversibility of H2 cleavage under these conditions was confirmed by using a H2/D2 mixture and observing that under these conditions no HD was generated.
- NMR spectral data for [tmpH][HB(C6F5)2(C6Cl5)] were reported in ref. 40 in CD3CN, we have re-measured this in CD2Cl2 to ensure the validity of comparisons with our data.
- H. Zaher, A. E. Ashley, M. Irwin, A. L. Thompson, M. J. Gutmann, T. Krämera and D. O'Hare, Chem. Commun., 2013, 49, 9755–9757 RSC.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- CrysAlisPro, Agilent Technologies, Yarnton, U.K Search PubMed.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 59–75 CrossRef CAS PubMed; O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS; L. Palatinus and A. van der Lee, J. Appl. Crystallogr., 2008, 41, 975–984 CrossRef; L. Palatinus, S. J. Prathapab and S. van Smaalen, J. Appl. Crystallogr., 2012, 45, 575–580 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef CAS PubMed; G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef PubMed.
- S. Lancaster, ChemSpider Synthetic Pages, 2003 Search PubMed , http://cssp.chemspider.com/215; T. E. Krafft, Boulder Scientific Company, US Pat. 5,679,289, 1997 CrossRef CAS PubMed; R. J. LeSuer, C. Buttolph and W. E. Geiger, Anal. Chem., 2004, 76, 6395–6401 CrossRef CAS PubMed.
- DigiElch-Professional, ElchSoft, Kleinromstedt, Germany Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further X-ray crystallographic data for 3; full characterisation data for Et3PO–3; further spectroscopic data (including time-resolved NMR spectra) for the FLP H2 cleavage reactions. CCDC 1446563. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra07007h |
| This journal is © The Royal Society of Chemistry 2016 |