
Blue light emitting self-healable graphene quantum dot embedded hydrogels†
Sagar Biswas,
Dnyaneshwar B. Rasale and
Apurba K. Das*
Discipline of Chemistry, Indian Institute of Technology Indore, Indore 452020, India. E-mail: apurba.das@iiti.ac.in
First published on 31st May 2016
Abstract
Graphene quantum dots (GQDs) embedded into synthesized Amoc (N-anthracenemethyloxycarbonyl) capped aromatic amino acid based units in completely aqueous environments exhibit blue light emitting self-healable hydrogels. These Amoc amino acid/GQD assemblies in hydrogel states encourage blue light emission under UV irradiation at a wavelength of 365 nm. The quenching in emission spectra reveals strong π–π stacking interactions within aromatic GQDs and Amoc-amino acids. Fluorescence spectra confirm the stabilization of GQDs on fibrils and electron microscopy images depict the distribution of GQDs on the nanofibrillar 3D networks. The self-healing properties and thixotropic nature of the hydrogels can be tuned by the inclusion of GQDs.
Introduction
Two dimensional graphene and graphene functionalized derivatives have attracted great attention due to their unique chemical and physical properties such as optical,1 electrochemiluminescent2 and cytotoxic behaviour.3 In addition, zero dimensional graphene quantum dots (GQDs) have also attracted researchers over the past few years due to their unique properties such as quantum confinement4 and edge effects.5 As a consequence, GQDs have been used in diverse research fields including photoconductivity,6 optoelectronics,7 bio-imaging,8 light emitting diodes,9 pH sensors10 and solar cell11 applications. Graphene functionalized materials have been used in bio-imaging8 and the enhancement of electrical conductivity.12 In nature, self-assembly is driven by hydrogen bonding, π–π stacking, hydrophobic and charge-transfer interactions.13 Graphene oxide (GO) embedded biomolecular hydrogels have important features including dye degradation capacity for wastewater treatment.14a A GO and poly-vinyl alcohol (PVA) composite produced a biocompatible hydrogel which was used for selective drug release application at physiological pH.14b A self-supported multifunctional hydrogel formed by GO-DNA composite via π-stacking interactions with nearly about 99 percent water content formed an excellent self-healing gel.15Peptides and amino acids are an interesting class of biomolecules, which can self-assemble to form hydrogels via non-covalent interactions.16 Supramolecular hydrogels were used in drug delivery,17 tissue engineering18 and different fields in biotechnology.19 Xu et al. reported peptide-hydrogel encapsulated hemin as an artificial enzyme to mimic peroxidase.20 Peptide and GO based hybrid hydrogel containing gold-nanoparticles was used as efficient catalyst to reduce aromatic nitro to aromatic amino groups.21 Recently, Wang et al. reported that self-assembled oligomers and carbon dots formed fluorescent ultra-long nanoribbons with electrical conductivity.22 Currently, self-healable materials are envisioned to serve as active materials in drug delivery23 and tissue engineering24 applications. In literature, there are limited reports on amino-acid/peptide-based thixotropic hydrogels.25 Self-healing materials can restore its original form by healing the damage caused on it26 and the healing process is driven by thermodynamic parameters27a–c and also depends on dynamic covalent bond27d–f and non-covalent28 interactions. Earlier reports give an account on peptide based self-healable multi-stimuli responsive metallo-hydrogel29 and automatic healing by polymer composite.30 Thixotropic and self-healing properties were also achieved from low-molecular-weight hydrogelators.31 Small bioactive molecules in presence of carbon based nanomaterials such as reduced graphene oxide (RGO)32,33 and single-walled carbon nanotubes (SWCNTs)34–38 were used to make hydrogels. It is also reported that SWCNTs weaken the thixotropic property of polysaccharide gels.39 Apart from that GQDs have completely different properties from other carbon based materials.40 Free amino acids are also used to prepare self-healing gels with GO and RGO.41 However, little attention has been paid on the properties of GQDs in presence of small aromatic functionalized molecules.22 To the best of our knowledge, there is no report on the effect of GQDs on gelation and influence on mechanical property of gels composed with N-caped amino acids. N-Protecting groups of amino acids and peptides have the ability to tune the mechanical and physical properties of supramolecular hydrogels.42a Single amino acid based hydrogels are limited in literature. Fmoc (9-fluorenylmethoxycarbonyl) protected amino acids and peptides were used for different biological application.42b In this work, our objective is to study the interaction between small aromatic functionalized biomolecules and GQDs. Herein; we demonstrate very simple N-terminal protected (Amoc: 9-anthracenemethyloxycarbonyl) amino acids (Amoc-F-OH and Amoc-Y-OH, F: L-phenylalanine; Y: L-tyrosine and –OH represents free acid group) which form self-healable hydrogels upon addition of GQDs at physiological pH 7.4. To study the interaction of small organic molecules with GQDs using spectroscopic techniques, anthracene moiety is used as good fluorophore. Aromatic amino acids helps to exhibit better gelation behaviour due to aromatic π–π stacking and hydrophobic interactions.42c Both aromatic amino acids phenylalanine and tyrosine have structural similarity and main difference is the presence of phenolic –OH group in p-position of tyrosine instead of having –H group in case of phenylalanine. Moreover, it is important to study the effect of phenolic –OH on the mechanical property of gels in presence of GQDs. Though there are several reports on self-healing gels embedded with graphene, GO32,43 and also with RGO but hydrogels of biomolecules embedded with GQDs are not studied extensively.44,45 GQDs exhibit solely different properties compare to other graphene based materials.40 So, it is important to study the behaviour of the embedded GQDs with biomolecules such as N-protected amino acids. The functionalized GQDs embedded hydrogels are found to exhibit blue light emission, which are analyzed by UV-Vis, fluorescence and electrochemical measurements. The morphological changes and self-healing properties of Amoc amino acids with inclusion of GQDs are also thoroughly assessed.
Results and discussion
Here, we have synthesized N-terminal protected (Amoc: 9-anthracenemethyloxycarbony) amino acids Amoc-F-OH and Amoc-Y-OH by the procedure46 described in Scheme 1. GQDs were synthesized by the simple hydrothermal process from commercially available GO and GQDs were purified by dialysis prior to gelation.47 We have prepared hydrogels with the compounds Amoc-F-OH (Gel-F), Amoc-Y-OH (Gel-Y) with 15 mmol L−1 of each amphiphilic molecules. We have added GQDs into the Amoc-F-OH (GQD-F) and Amoc-Y-OH (GQD-Y) to tune the mechanical property of the gels (Table 1) and concentration of GQDs was optimized using fluorescence spectroscopic technique. | ||
| Scheme 1 General synthetic scheme of gelators 1 and 2. | ||
| Gelator | Conc. of gelator (mmol L−1) | Amount of GQDs (mg mL−1) | Property of Gel |
|---|---|---|---|
| Amoc-F-OH | 15 | 0 | Gel |
| 10 | 0 | Sol | |
| 10 | 0.2 | Gel | |
| 10 | 0.5 | Self-healing gel | |
| 7.5 | 0.2 | Gel | |
| 7.5 | 0.5 | Gel | |
| Amoc-Y-OH | 15 | 0 | Gel |
| 10 | 0 | Sol | |
| 10 | 0.2 | Gel | |
| 10 | 0.5 | Self-healing gel | |
| 7.5 | 0.2 | Gel | |
| 7.5 | 0.5 | Gel |
Different concentration of Amoc-F-OH/Amoc-Y-OH and GQDs were used to prepare hydrogels (Table 1). However, self-healable hydrogels were formed (Fig. 1) upon mixing of 10 mmol L−1 Amoc-Y-OH/Amoc-Y-OH and 0.5 mg mL−1 GQDs (Table 1). These amphiphilic molecules formed gels using hydrophobic-hydrophilic and other non-covalent interactions at physiological pH.48 Hence, gels were prepared at physiological condition (pH = 7.4) and gelation was initially confirmed by test tube inversion method. For the preparation of self-healable hydrogel GQD-F, 0.449 wt% of Amoc-F-OH and GQDs was used. 0.466 wt% Amoc-Y-OH and GQDs was used for the preparation of hydrogel GQD-Y. Both the cases water content was more than 99.5%. At higher pH (>7.5), the gelator compounds start to dissolve in the water medium and give clear solution of gelator molecules at pH > 8. But at lower pH (pH < 6), the gelator molecules get precipitate from the solution.
 | ||
| Fig. 1 Schematic representation of gel nanofibers and GQDs embedded gel nanofibers through molecular self-assembly. | ||
Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) images (ESI Fig. S1†) reveal the size of 7–8 nm of GQDs according to size distribution graph, which was acquired from TEM image (ESI Fig. S2†). SEM images of Gel-F and Gel-Y exhibit fibrillar networks (ESI Fig. S3†). The SEM images (ESI Fig. S3†) of GQD-F and GQD-Y are consistent with the TEM images (Fig. 2) of GQD-F and GQD-Y. Both the cases (GQD-F and GQD-Y), GQDs are decorated on the fibers and the average width of the nanofibrillar network is around 20 nm (Fig. 2 and ESI Fig. S4†). GQD-Y shows straight nanofibrillar network where as GQD-F exhibits nanofibrillar ribbon with sheet type network observed from SEM image (ESI,† Fig. 3c). The morphological difference is attributed to the structural change of gelators. Different concentration of GQDs was used to observe the change in GQDs decorated nanofibrils. TEM images of GQD-F and GQD-Y (Amoc-F-OH/Amoc-Y-OH ≡ 10 mmol L−1 with 0.3 mg L−1 GQDs) exhibit nanofibrils decorated with a very less amount of GQDs (Fig. S5a and c†). TEM images of GQD-F and GQD-Y (Amoc-F-OH/Amoc-Y-OH ≡ 10 mmol L−1 with 0.7 mg L−1 GQDs) reveal nanofibrils decorated with GQDs (ESI Fig. S5b and d†) which look similar to the distribution of GQDs on nanofibrils (Fig. 2).
 | ||
| Fig. 2 TEM images of (a) GQD-F (10 mmol L−1 of Amoc-F-OH with 0.5 mg L−1 GQDs) and (b) GQD-Y (10 mmol L−1 of Amoc-Y-OH with 0.5 mg L−1 GQDs) show GQDs decorated nanofibrillar structures. Both images depict uniform distribution of GQDs on the fibrillar networks. | ||
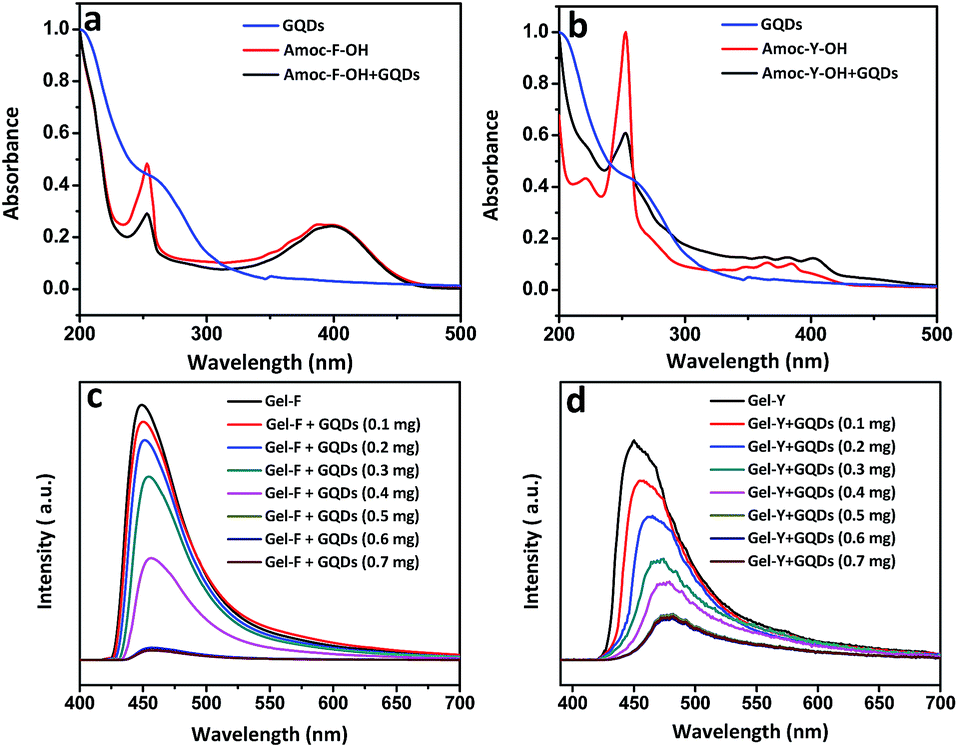 | ||
| Fig. 3 (a) and (b) show UV-Vis spectra of GQDs, Gel-F, Gel-Y, GQD-F and GQD-Y. Red shift in UV-Vis was observed for GQD-Y. Fluorescence spectra of (c) Gel-F (10 mmol L−1), GQD-F and (d) Gel-Y (10 mmol L−1), GQD-Y show successive quenching on increasing addition of GQDs. | ||
UV-Vis absorption spectra of GQDs depict π–π* transition at 206 nm for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and also n–π* transition at 260 nm due to the functionalized CO group (Fig. 3a and b).49 An absorption band at 251 nm is attributed to the presence of phenylalanine moiety. A broad peak in the region of 380–410 nm was observed for both Gel-F and GQD-F. Absorption spectra of both Gel-Y and GQD-Y show absorption band at 254 nm and similar red shift was observed from 380 nm to 385 nm due to the π-stacking interaction of Amoc group and GQDs (Fig. 3b). Fluorescence spectra were acquired to know about more insight of GQDs and GQDs embedded Amoc-amino acid based hydrogels (Fig. 3c and d). GQDs show excitation wavelength dependent photoluminescence spectra (ESI Fig. S6†).50 After the inclusion of GQDs into Amoc-amino acids based hydrogels (GQD-F and GQD-Y), we acquired photoluminescence spectra of Amoc-F-OH and GQD-F at an excitation wavelength of 390 nm and Amoc-Y-OH and GQD-Y at an excitation wavelength of 380 nm (Fig. 3a and b). Both the gels (GQD-F and GQD-Y) imparted successive fluorescence quenching as compare to the Amoc-F/Y-OH without GQDs. The fluorescence quenching (Fig. 3c and d) indicates strong π–π stacking interactions between aromatic anthracene moieties and GQDs.51,52 Before the inclusion of GQDs, intense emission peak appeared at 450 nm for both Amoc-F-OH and Amoc-Y-OH. With increasing addition of GQDs into Amoc amino acids, successive fluorescence quenching was observed with red shift. Concentration dependent emission spectra of 10 mmol L−1 of gelators (Amoc-F-OH/Amoc-Y-OH) were recorded with the varied concentration of GQDs (0 to 0.7 mg L−1). The emission intensity gradually decreases with the increase concentration of GQDs up to 0.5 mg mL−1 for both the gels GQD-F and GQD-Y. However, there is no significant change in emission wavelength and intensity after varying the concentration of GQDs from 0.5 mg L−1 to 0.7 mg L−1. This result signifies that, particular concentration of GQDs (0.5 mg L−1) is required for a particular concentration of gelators (10 mmol L−1) to achieve maximum interaction with GQDs and the formation of self-healing gel. GQD-F shows quenched emission maxima at 464 nm where as GQD-Y exhibits emission maxima at 480 nm after fluorescence quenching. The red shift signifies stabilization of GQDs on the fibers by non-covalent interaction including π–π stacking interaction. The shift is different for two different gels which is attributed to the change in the structural unit of the gelator molecules. The phenolic –OH group of Amoc-Y-OH has affinity over the functionalized GQDs which helps edge-localized π–π stacking interaction between Amoc-Y-OH and GQDs. This interaction promotes the composite to shift more towards higher wavelength.53
C bond and also n–π* transition at 260 nm due to the functionalized CO group (Fig. 3a and b).49 An absorption band at 251 nm is attributed to the presence of phenylalanine moiety. A broad peak in the region of 380–410 nm was observed for both Gel-F and GQD-F. Absorption spectra of both Gel-Y and GQD-Y show absorption band at 254 nm and similar red shift was observed from 380 nm to 385 nm due to the π-stacking interaction of Amoc group and GQDs (Fig. 3b). Fluorescence spectra were acquired to know about more insight of GQDs and GQDs embedded Amoc-amino acid based hydrogels (Fig. 3c and d). GQDs show excitation wavelength dependent photoluminescence spectra (ESI Fig. S6†).50 After the inclusion of GQDs into Amoc-amino acids based hydrogels (GQD-F and GQD-Y), we acquired photoluminescence spectra of Amoc-F-OH and GQD-F at an excitation wavelength of 390 nm and Amoc-Y-OH and GQD-Y at an excitation wavelength of 380 nm (Fig. 3a and b). Both the gels (GQD-F and GQD-Y) imparted successive fluorescence quenching as compare to the Amoc-F/Y-OH without GQDs. The fluorescence quenching (Fig. 3c and d) indicates strong π–π stacking interactions between aromatic anthracene moieties and GQDs.51,52 Before the inclusion of GQDs, intense emission peak appeared at 450 nm for both Amoc-F-OH and Amoc-Y-OH. With increasing addition of GQDs into Amoc amino acids, successive fluorescence quenching was observed with red shift. Concentration dependent emission spectra of 10 mmol L−1 of gelators (Amoc-F-OH/Amoc-Y-OH) were recorded with the varied concentration of GQDs (0 to 0.7 mg L−1). The emission intensity gradually decreases with the increase concentration of GQDs up to 0.5 mg mL−1 for both the gels GQD-F and GQD-Y. However, there is no significant change in emission wavelength and intensity after varying the concentration of GQDs from 0.5 mg L−1 to 0.7 mg L−1. This result signifies that, particular concentration of GQDs (0.5 mg L−1) is required for a particular concentration of gelators (10 mmol L−1) to achieve maximum interaction with GQDs and the formation of self-healing gel. GQD-F shows quenched emission maxima at 464 nm where as GQD-Y exhibits emission maxima at 480 nm after fluorescence quenching. The red shift signifies stabilization of GQDs on the fibers by non-covalent interaction including π–π stacking interaction. The shift is different for two different gels which is attributed to the change in the structural unit of the gelator molecules. The phenolic –OH group of Amoc-Y-OH has affinity over the functionalized GQDs which helps edge-localized π–π stacking interaction between Amoc-Y-OH and GQDs. This interaction promotes the composite to shift more towards higher wavelength.53
GQDs exhibit semiconducting property54 and the study of electrochemical property after inclusion of GQDs into gelator molecules is important. Cyclic voltammetry shows only a reduction peak at a potential value of −0.867 (V) with quasi-reversibility in case of GQDs but both Gel-F and Gel-Y show reversible potential curves (Fig. 4). Cyclic voltammetry study depicts reversible graph for GQD-F with a weak oxidation peak at 0.9704 V (Fig. 4a). GQD-Y also shows reversible graph with strong oxidation peak at 1.099 V (Fig. 4b). After inclusion of GQDs into the Amoc-amino acids, electron transfer occurs from GQDs to anthracene moieties for both the cases and the resultant composites act as semiconductors.
 | ||
| Fig. 4 (a) and (b) represent the comparison of cyclic voltammetry data of GQDs, Gel-F, GQD-F (10 mmol L−1 of Amoc-F-OH with 0.5 mg L−1 GQDs) and GQDs, Gel-Y, GQD-Y (10 mmol L−1 of Amoc-Y-OH with 0.5 mg L−1 GQDs) respectively. | ||
Rheological experiment reveals more insight about the mechanical and thixotropic properties of hydrogels. Rheological experiments were performed with self-healable hydrogels GQD-F (10 mmol L−1 of Amoc-F-OH with 0.5 mg L−1 GQDs) and GQD-Y (10 mmol L−1 of Amoc-Y-OH with 0.5 mg L−1 GQDs). In the oscillatory frequency sweep measurements, all the samples show higher storage modulus (G′) value with compare to loss modulus (G′′) over the entire process (Fig. 5a and 6a). Higher storage modulus compare to loss modulus supports the formation of rigid gels.55 G′ value increases from 160 Pa to 1180 Pa (shown in the plot of error bar ESI Fig. S7†) after the inclusion of GQDs into the Amoc-F-OH and in the other case G′ increases from 213 Pa (Gel-Y) to 710 Pa (ESI Fig. S7†) for GQD-Y (Fig. 5a and 6a). After inclusion of GQDs into hydrogel Gel-F, the hydrogel GQD-F becomes stronger than Gel-F. Similarly GQD-Y becomes stronger than hydrogel Gel-Y. A step strain experiment was performed at constant angular frequency of 10 rad s−1 to quantify the thixotropic properties of hydrogels (Fig. 5b and 6b). A constant strain of 0.1% was applied (step 1) to hydrogels GQD-F and GQD-Y. The fibrillar 3D network completely ruptured when the strain is slowly increased from 0.1% to 20% (step 2) and it took 5 minutes to deliver gel to sol transition (G′ < G′′). Sol to gel transition (G′ > G′′) was observed by applying low strain to 0.1% (step 3) upto 6 min. The storage modulus (G′) of GQD-F restored to its present position after 6 min (Fig. 5b) due to reformation of fibrillar 3D networks.56 But the storage modulus of GQD-Y was 710 Pa before applying the strain. GQD-Y recovered upto 98% after removing the strain (step 3). After applying 20% strain (step 4), both the gels attain gel-to-sol property (G′ < G′′). Finally after reduction of strain from 20% to 0.1%, the storage modulus (G′) was restored to its present position for GQD-F. However, the storage modulus (G′) was restored upto 84% for GQD-Y. Hydrophobicity (expressed as the partition coefficient between water and octanol, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) of Amoc-F-OH (logP = 5.22) and Amoc-Y-OH (logP = 4.74) is different due to structural difference. Hydrophobicity of the gelator can also tune the mechanical property of the gel57 as a result of this, the gel recovery property becomes different for GQD-F and GQD-Y. Due to enhanced non-covalent interaction between gelators and GQDs including π–π stacking interaction, the resultant gels become more thixotropic which provide better self-healing property. Though both the above results give an account for the thixotropic nature of the GQDs embedded hydrogel.
P) of Amoc-F-OH (logP = 5.22) and Amoc-Y-OH (logP = 4.74) is different due to structural difference. Hydrophobicity of the gelator can also tune the mechanical property of the gel57 as a result of this, the gel recovery property becomes different for GQD-F and GQD-Y. Due to enhanced non-covalent interaction between gelators and GQDs including π–π stacking interaction, the resultant gels become more thixotropic which provide better self-healing property. Though both the above results give an account for the thixotropic nature of the GQDs embedded hydrogel.
 | ||
| Fig. 5 Dynamic frequency sweep experiment: (a) GQD-F (10 mmol L−1 of Amoc-F-OH with 0.5 mg L−1 GQDs) becomes 6 times more rigid than Gel-F due to interaction of GQDs. (b) Step strain experiment signifies thixotropic nature and excellent gel recovery of GQD-F. | ||
 | ||
| Fig. 6 (a) GQD-Y (10 mmol L−1 of Amoc-Y-OH with 0.5 mg L−1 GQDs) becomes two times rigid than Gel-Y after inclusion of GQDs represented in dynamic frequency sweep experiment. (b) Step strain experiment describes thixotropic nature and self-healing property of the gel GQD-Y. | ||
Autonomous healing upon damage on material is recognized as self-healing property.58,59 This fascinating property was observed during the experiments with the composite of Amoc-amino acids and GQDs (Fig. 7 and ESI Fig. S8†). To quantify the self-healing properties of hydrogels, we cut the gel GQD-F with knife into two pieces (Fig. 7f) and shoved to keep contact with each other for a minute (Fig. 7g). These two pieces stuck together into a strong self-healable gel which was capable to make a bridge with two vial caps (Fig. 7g). Later, we cut the self-healable gel into four small pieces and adhered together to make a self-healable gel (Fig. 7i). Supramolecular interaction such as π–π stacking, hydrophobic and hydrogen bonding interaction59 (Fig. 1) are responsible for the dynamic property of the formed cross-linking fibrillar networks in gels. Self-healing property of small amphiphilic molecules is driven by the thermodynamic parameter i.e. entropy. Entropy increases due to the incremental interfacial area with cracking the material by applying the external mechanical forces. With the deformation of the gel composed by small molecules, the number of arrangement increases with increasing entropy in that particular state.27a–c Increased entropy drives the gelator molecules to come closer to the damaged space by extending the crosslinked 3D network which leads to the formation of the previous shape.
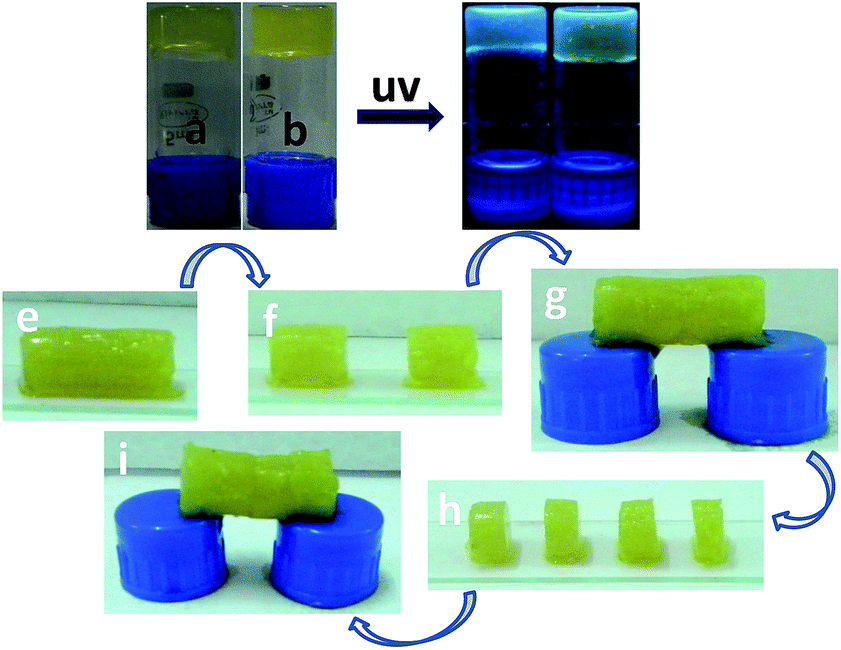 | ||
| Fig. 7 (a) and (b) are the optical images of GQD-F, GQD-Y respectively under day light. (c) and (d) are the images under UV light irradiation at a wavelength of 365 nm of hydrogels GQD-F (10 mmol L−1 of Amoc-F-OH with 0.5 mg L−1 GQDs) and GQD-Y (10 mmol L−1 of Amoc-Y-OH with 0.5 mg L−1 GQDs) respectively. Optical images (e) to (i) represent the self-healing property of GQD-F (10 mmol L−1 of Amoc-F-OH with 0.5 mg L−1 GQDs). Hydrogel pieces adhere together after keep in contact with each other and regenerate previous shape by crosslinking network by noncovalent interaction. | ||
Conclusions
We have demonstrated self-healable GQDs embedded Amoc capped aromatic amino acid based hydrogels at physiological pH. GQDs/Amoc-amino acid based hybrid hydrogels emit blue light under UV light irradiation at 365 nm. Aromatic Amoc amino acids based hydrogels formed nanofibrillar networks. Fluorescence spectroscopic studies of GQDs embedded hydrogels reveal that GQDs are stabilized by nanofibrillar networks. Morphological changes of GQD-F and GQD-Y were observed due to the change in gelator structure and the stabilization of GQDs in the 3D network of gels. Hydroxyl group in tyrosine affects significantly to the mechanical property of the hydrogel. Aromatic π–π stacking interactions within GQDs and Amoc amino acids and hydrogen bonding interactions are the driving force to stabilize the GQDs by the nanofibrillar networks. Fluorescence quenching reveals strong π–π stacking interactions51,52 within GQDs and Amoc-amino acids. Electron transfer from GQDs to anthracene moieties is studied using electrochemical analysis and the resultant composites act as semiconductor. GQDs embedded Amoc-amino acids based hydrogels afford thixotropic in nature. The hydrogel composites of Amoc-F-OH and GQDs deliver excellent entropy driven self-healable behaviour, which can be used in drug delivery, tissue engineering and cell damage repairing in biological systems in future.Experimental
General methods
All the chemicals and reagents were obtained commercially. All NMR spectra were recorded with 400 MHz Bruker AV 400 NMR. Compounds concentrations were in the range of 1–10 mmol L−1 in (CD3)2SO and CDCl3. Mass spectra were recorded on Bruker micrOTOF-Q II by positive electrospray ionisations. All the reported FT-IR spectra were taken using Bruker (Tensor 27) FT-IR spectrophotometer. Specific rotations of the synthesized compounds were measured on an AutopolR V automatic polarimeter (Rudolph research analytical). The cell (length = 100 mm, capacity = 2 mL) was used for this study at 25 °C.Synthesis of anthracen-9-ylmethyl (4-nitrophenyl) carbonate 3 (ref. 37)
A stirred solution of p-nitrochloroformate (1.259 g, 6.25 mmol) in DCM was cooled in ice bath under argon atmosphere. Dimethylaminopyridine (DMAP) (0.65 g, 5.28 mmol) was added under the same reaction condition. After addition of DMAP, a white slurry was obtained. 9-Anthracenemethanol (1 g, 4.8 mmol) was added to the reaction mixture by several portions. The reaction mixture was allowed to stir overnight at room temperature. TLC showed complete conversion of product. After that DCM was evaporated to dryness in a rotary-evaporator. The crude reaction mixture was diluted with ethyl acetate (50 mL). The mixture was washed with 0.5 M HCl (3 × 50 mL) and successively with brine. Light yellow mass was obtained by evaporating the solvent under reduced pressure and was crystallized from benzene.Yield = 1.6 g (4.28 mmol, 89%); 1H NMR (400 MHz, CDCl3): δ 8.49 (s, 1H, C8–H of Anth), 8.34–8.32 (d, 2H, J = 8.76, Ar-Hs), 8.17–8.15 (d, 2H, J = 9.04, Ar-Hs), 7.99–7.97 (d, 2H, J = 8.52, Ar-Hs), 7.56–7.53 (t, 2H, Ar-Hs), 7.46–7.43 (t, 2H, Ar-Hs), 7.29–7.26 (d, 2H, J = 9.04, Ar-Hs), 6.30 (s, 2H, −CH2) (ESI Fig. S10†).
Preparation of Amoc-Phe-OH 1 (ref. 37)
1.5 g (4.02 mmol) of anthracen-9-ylmethyl (4-nitrophenyl) carbonate 3 was dissolved in 3 mL DMF in a 100 mL round bottom flask and cooled it in an ice bath. A neutralized solution of phenylalanine methyl ester was extracted from its corresponding hydrochloride salt (1.73 g, 8.04 mmol) and concentrated to add to the reaction mixture. The progress of the reaction was monitored by thin layer chromatography (TLC). The reaction mixture was allowed to stir for 8 h. The reaction mixture was diluted with ethyl acetate and washed with 1 (N) HCl (3 × 50 mL), saturated Na2CO3 solution (3 × 50 mL) and then with brine. Solid yellow mass was obtained after evaporating the solvent under reduced pressure.Without purification, a solution of Amoc-Phe-OMe 4 in 5 mL distilled dry methanol was allowed to react with a solution of 5 mL 1 (N) NaOH solution. The reaction progress was monitored by thin layer chromatography. The reaction mixture was stirred upto 4 h. After the completion of the reaction, excess methanol was evaporated to dryness and diluted with 100 mL water. Then the water mixture was taken in a separating funnel and vigorously washed with diethyl ether. The aqueous layer was collected and cooled in an ice bath. Then, the cooled collected water layer was acidified with 1 (N) KHSO4. The pH of aqueous layer was adjusted to 2 and the product was extracted with ethyl acetate (3 × 30 mL). The ethyl acetate layer was dried over anhydrous sodium sulphate and evaporated under reduced pressure to obtain orange-yellow solid compound Amoc-Phe-OH 1.37
Yield 1.47 g (3.68 mmol, 92%); [α]25D = −0.067 (c = 1, MeOH); FT-IR (KBr): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) 3312 (br), 3029 (br), 1688 (s), 1599 (s), 1510 (s), 1441 (s), 1258 (s); 1H NMR (400 MHz, DMSO-d6): δ 8.60 (s, 1H, C8–H of Anth), 8.27–8.24 (d, 2H, J = 8.52, Ar-Hs), 8.06–8.04 (d, 2H, 8.28, Ar-Hs), 7.54–7.46 (m, 5H, Ar-Hs), 7.17 (s, 4H, Ar-Hs), 5.99–5.88 (m, 2H, −CH2 of Amoc), 4.17–4.12 (m, 1H, CαH of Phe), 2.99–2.95 (m, 1H, CβH of Phe), 2.75–2.69 (m, 1H, CβH of Phe) (ESI Fig. S11†). 13C NMR (100 MHz, DMSO-d6): δ 173.32, 156.20, 137.86, 130.85, 130.41, 129, 128.09, 126.58, 125.22, 124.13, 58.04, 55.59, 36.32 (ESI Fig. S12†). MS (ESI) m/z for C25H21NO4 (M + H)+ calcd: 422.1363, found: 422.1385 (ESI Fig. S13†).
3312 (br), 3029 (br), 1688 (s), 1599 (s), 1510 (s), 1441 (s), 1258 (s); 1H NMR (400 MHz, DMSO-d6): δ 8.60 (s, 1H, C8–H of Anth), 8.27–8.24 (d, 2H, J = 8.52, Ar-Hs), 8.06–8.04 (d, 2H, 8.28, Ar-Hs), 7.54–7.46 (m, 5H, Ar-Hs), 7.17 (s, 4H, Ar-Hs), 5.99–5.88 (m, 2H, −CH2 of Amoc), 4.17–4.12 (m, 1H, CαH of Phe), 2.99–2.95 (m, 1H, CβH of Phe), 2.75–2.69 (m, 1H, CβH of Phe) (ESI Fig. S11†). 13C NMR (100 MHz, DMSO-d6): δ 173.32, 156.20, 137.86, 130.85, 130.41, 129, 128.09, 126.58, 125.22, 124.13, 58.04, 55.59, 36.32 (ESI Fig. S12†). MS (ESI) m/z for C25H21NO4 (M + H)+ calcd: 422.1363, found: 422.1385 (ESI Fig. S13†).
Compound Amoc-Tyr-OH 2 was also synthesized accordingly.37 Yield = 0.97 g, (2.34 mmol, 91%), [α]25D = −2.9 (c = 1, MeOH); FT-IR (KBr): 3416 (br), 3198 (br), 1592 (s), 1605 (s), 1510 (s), 1454 (s), 1333 (s); 1H NMR (400 MHz, DMSO-d6): δ 9.08 (s, 1H), 8.53 (s, 1H, C8–H of Anth), 8.19–8.17 (d, 2H, J = 8.76, Ar-Hs), 7.99–7.97 (d, 2H, J = 8.04, Ar-Hs), 7.47–7.38 (m, 4H, Ar-Hs), 7.31–7.29 (d, 1H, J = 8.04, Ar-H), 6.86–6.83 (d, 2H, J = 8.28, Ar-Hs), 6.48–6.46 (d, 2H, J = 8.04, Ar-Hs), 5.92–5.80 (m, 2H, −CH2 of Amoc), 4.01–3.95 (m, 1H, CαH of Tyr), 2.79–2.74 (m, 1H, CβH of Tyr), 2.56–2.50 (m, 1H, CβH of Tyr) (ESI Fig. S14†). 13C NMR (100 MHz, DMSO-d6): δ 173.4, 156.18, 155.79, 130.86, 130.42, 129.93, 128.86, 126.59, 125.22, 124.12, 114.9, 58.06, 55.94, 35.64 (ESI Fig. S15†). MS (ESI) m/z for C25H21NO5 (M + H)+ calcd: 438.1312, found: 438.1333 (ESI Fig. S16†).
Synthesis of graphene quantum dots (GQDs)39
Commercially available GO (800 mg) was heated in a furnace at 200 °C for two hours to afford graphene sheet and the resultant graphene sheets were mixed with concentrated 40 mL of (3:1) HNO3:H2SO4 mixture. The mixture was heated at 90 °C for 3 h and it was diluted with 250 mL distilled water. Then the mixture was filtered through 0.22 μm microporous syringe filter to remove the acids. Then, the purified (300 mg) oxidized graphene sheets (GSs) were dissolved in 50 mL of millipore water and the pH was adjusted to 8 with NaOH. The mixture was taken in a reaction bomb vessel. Then the reaction bomb vessel was kept into a hot air oven for 12 hours of constant heating at 200 °C. Finally, the solution was dialyzed for 24 hours with a dialysis tube of 3500 K Dalton to remove ions and impurity from solution. The formation of GQDs was confirmed by IR spectroscopy (ESI Fig. S9†), SEM and TEM images (ESI Fig. S1†).
Preparation of hydrogels
Morphological study
Transmission electron microscopic images were taken using a JEOL electron microscope (model: JEM-2100), operated at an accelerating voltage of 200 kV and Field Emission Gun-Transmission Electron Microscope (model: Tecnai G2, F30), operated on a voltage of 300 kV. 100 μL of gel was dissolved in 200 μL of water and the dilute solution of the hydrogels was dried on carbon-coated copper grids (300 mesh) by slow evaporation in air and then allowed to dry separately in a vacuum at room temperature. GQDs solution in DI water also dried on carbon-coated copper grids. Field emission scanning electron microscope (FE-SEM Supra 55 Zeiss) instrument was used for SEM study. Gels were dried on cover slip and coated with gold for SEM analysis with an operating voltage of 5 kV.UV-Vis spectroscopy
UV-Vis absorption spectra of the hydrogels Gel-F, Gel-Y, GQD-F and GQD-Y were recorded using a Varian Cary100 Bio UV-Vis spectrophotometer at a concentration of 5 mmol L−1. UV-Vis absorption spectra of GQD were recorded at a concentration of 0.25 g L−1.Fluorescence spectroscopy
Fluorescence emission spectra of hydrogels (10 mmol L−1) were recorded at different excitation wavelength of 390 nm and 380 nm with medium sensitivity on a Horiba Scientific Fluoromax-4 spectrophotometer. The slit width for the excitation and emission was set at 2 nm and a 1 nm data pitch. Samples were prepared in 1 cm2 quartz cuvette at room temperature.Cyclic voltammetry study
Electrochemical analysis was performed using CHI620D electrochemical analyzer. Glassy carbon was used as working electrode, platinum was used as counter electrode and Ag/AgCl was used as the reference electrode. Concentration of the gel samples was 10 mmol L−1 and the electrolyte KCl (0.1 mmol L−1) was used.Rheological study
Rheological study was performed to determine the mechanical properties of hydrogels. These properties were assessed using an Anton Paar Physica Rheometer (MCR 301, Austria) with parallel plate geometry (25 mm in diameter) and temperature was controlled at 25 °C. The dynamic moduli of the hydrogel were measured as a function of frequency in the range of 0.1–100 rad s−1 with a constant strain value 0.1% and step strain experiment was done at the constant frequency of 10 rad s−1 and applied strain was changed from 0.1% to 20%. 200 μL of gel was prepared in glass vial and transferred it over the plate using micro-spatula to proceed for rheological measurements. To check the reproducibility, each experiment was carried out for three times for average value was taken for discussion. Storage and loss modulus in rheological experiments60 were plotted with error bars (Fig. S7†).Acknowledgements
This work was supported by IIT Indore and Department of Science & Technology (Project No. SR/NM/NS-1458/2014), New Delhi, India. SB is indebted to UGC, New Delhi for his fellowship. DBR is thankful to the IIT Indore for a postdoctoral fellowship. We thank SAIF, NEHU, Shillong and SAIF, IIT Bombay for the assistance of TEM facility. The sophisticated instrumentation centre (SIC), IIT Indore is acknowledged for providing access to the instrumentation.References
- W. Kwon, Y.-H. Kim, C.-L. Lee, M. Lee, H.-C. Choi, T.-W. Lee and S.-W. Rhee, Nano Lett., 2014, 14, 1306–1311 CrossRef CAS PubMed.
- (a) L. Y. Zheng, Y. W. Chi, Y. Q. Dong, J. P. Lin and B. B. Wang, J. Am. Chem. Soc., 2009, 131, 4564–4565 CrossRef CAS PubMed; (b) Y. Dong, W. Tian, S. Ren, R. Dai, Y. Chi and G. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 1646–1651 CrossRef CAS PubMed.
- (a) S. Zhu, J. Zhang, S. Tang, C. Qiao, L. Wang, H. Wang, X. Liu, B. Li, Y. Li, W. Yu, X. Wang, H. Sun and B. Yang, Adv. Funct. Mater., 2012, 22, 4732–4740 CrossRef CAS; (b) S. Zhu, J. Zhang, C. Qiao, S. Tang, Y. Li, W. Yuan, B. Li, L. Tian, F. Liu, R. Hu, H. Gao, H. Wei, H. Zhang, H. Sun and B. Yang, Chem. Commun., 2011, 47, 6858–6860 RSC; (c) Y. Dong, C. Chen, X. Zheng, L. Gao, Z. Cui, H. Yang, C. Guo, Y. Chi and C. M. Li, J. Mater. Chem., 2012, 22, 8764–8766 RSC.
- (a) S. Zhuo, M. Shao and S. T. Lee, ACS Nano, 2012, 6, 1059–1064 CrossRef CAS PubMed; (b) X. Yan, B. Li, X. Cui, Q. Wei, K. Tajima and L. S. Li, J. Phys. Chem. Lett., 2011, 2, 1119–1124 CrossRef CAS PubMed.
- (a) J. Shen, Y. Zhu, C. Chen, X. Yang and C. Li, Chem. Commun., 2011, 47, 2580–2582 RSC; (b) R. Liu, D. Wu, X. Feng and K. Muellen, J. Am. Chem. Soc., 2011, 133, 15221–15223 CrossRef CAS PubMed.
- (a) G. Konstantatos, M. Badioli, L. Gaudreau, J. Osmond, M. Bernechea, P. G. F. de Arquer, F. Gatti and F. H. L. Koppens, Nat. Nanotechnol., 2012, 7, 363–368 CrossRef CAS PubMed; (b) V. Gupta, N. Chaudhary, R. Srivastava, G. D. Sharma, R. Bhardwaj and S. Chand, J. Am. Chem. Soc., 2011, 133, 9960–9963 CrossRef CAS PubMed; (c) P. Routh, S. Das, A. Shit, P. Bairi, P. Das and A. K. Nandi, ACS Appl. Mater. Interfaces, 2013, 5, 12672–12680 CrossRef CAS PubMed.
- F. Bonaccorso, Z. Sun, T. Hasan and A. C. Ferrari, Nat. Photonics, 2010, 4, 611–622 CrossRef CAS.
- M. Nurunnabi, Z. Khatun, K. M. Huh, S. Y. Park, D. Y. Lee, K. J. Cho and Y.-K. Lee, ACS Nano, 2013, 7, 6858–6867 CrossRef CAS PubMed.
- D. I. Son, B. W. Kwon, D. H. Park, W.-S. Seo, Y. Yi, B. Angadi, C.-L. Lee and W. K. Choi, Nat. Nanotechnol., 2012, 7, 465–471 CrossRef CAS PubMed.
- K. Paek, H. Yang, J. Lee, J. Park and B. J. Kim, ACS Nano, 2014, 8, 2848–2856 CrossRef CAS PubMed.
- J. K. Kim, M. J. Park, S. J. Kim, D. H. Wang, S. P. Cho, S. Bae, J. H. Park and B. H. Hong, ACS Nano, 2013, 7, 7207–7212 CrossRef CAS PubMed.
- (a) G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS PubMed; (b) M. A. Worsley, P. J. Pauzauskie, T. Y. Olson, J. Biener, J. H. Satcher and T. F. Baumann, J. Am. Chem. Soc., 2010, 132, 14067–14069 CrossRef CAS PubMed.
- (a) S. K. M. Nalluri, C. Berdugo, N. Javid, P. W. J. M. Frederix and R. V. Ulijn, Angew. Chem., Int. Ed., 2014, 53, 5882–5887 CrossRef CAS PubMed; (b) J. M. Poolman, J. Boekhoven, A. Besselink, A. G. L. Olive, J. H. van Esch and R. Eelkema, Nat. Protoc., 2014, 9, 977–988 CrossRef CAS PubMed.
- (a) T. Jiao, H. Zhao, J. Zhou, Q. Zhang, X. Luo, J. Hu, Q. Peng and X. Yan, ACS Sustainable Chem. Eng., 2015, 3, 3130–3139 CrossRef CAS; (b) H. Bai, C. Li, X. Wang and G. Shi, Chem. Commun., 2010, 46, 2376–2378 RSC.
- Y. Xu, Q. Wu, Y. Sun, H. Bai and G. Shi, ACS Nano, 2010, 4, 7358–7362 CrossRef CAS PubMed.
- (a) X. Yan, Y. Cui, Q. He, K. Wang and J. Li, Chem. Mater., 2008, 20, 1522–1526 CrossRef CAS; (b) I. Maity, D. B. Rasale and A. K. Das, Soft Matter, 2012, 8, 5301–5308 RSC; (c) D. B. Rasale, I. Maity and A. K. Das, RSC Adv., 2012, 2, 9791–9794 RSC; (d) Q. Zou, L. Zhang, X. Yan, A. Wang, G. Ma, J. Li, H. Mçhwald and S. Mann, Angew. Chem., Int. Ed., 2014, 53, 2366–2370 CrossRef CAS PubMed.
- S. Bhuniya, S. M. Park and B. H. Kim, Org. Lett., 2005, 7, 1741–1744 CrossRef CAS PubMed.
- M.-H. Yao, J. Yang, J.-T. Song, D.-H. Zhao, M.-S. Du, Y.-D. Zhao and B. Liu, Chem. Commun., 2014, 50, 9405–9408 RSC.
- K. M. Galler, L. Aulisa, K. R. Regan, R. N. D'Souza and J. D. Hartgerink, J. Am. Chem. Soc., 2010, 132, 3217–3223 CrossRef CAS PubMed.
- Q. Wang, Z. Yang, X. Zhang, X. Xiao, C. K. Chang and B. Xu, Angew. Chem., Int. Ed., 2007, 46, 4285–4289 CrossRef CAS PubMed.
- B. Adhikari, A. Biswas and A. Banerjee, ACS Appl. Mater. Interfaces, 2012, 4, 5472–5482 CAS.
- G. Zhang, M. Yan, X. Teng, H. Bi, Y. Han, M. Tian and M. Wang, Chem. Commun., 2014, 50, 10244–10247 RSC.
- (a) Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS; (b) J. M. Knipe and N. A. Peppas, Regen. Biomater, 2014, 1, 57–65 CrossRef PubMed; (c) G. Chang, Y. Chen, Y. Li, S. Li, F. Huang, Y. Shen and A. Xie, Carbohydr. Polym., 2015, 122, 336–342 CrossRef CAS PubMed.
- (a) K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1879 CrossRef CAS PubMed; (b) J. Zhu and R. E. Marchant, Expert Rev. Med. Devices, 2011, 8, 607–626 CrossRef CAS PubMed; (c) C. T. S. W. P. Foo, J. S. Lee, W. Mulyasasmita, A. Parisi-Amon and S. C. Heilshorn, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22067–22072 CrossRef CAS PubMed.
- (a) J. Nanda, A. Biswas and A. Banerjee, Soft Matter, 2013, 9, 4198–4208 RSC; (b) H. Liu, Y. Hu, H. Wang, J. Wang, D. Kong, L. Wang, L. Chen and Z. Yang, Soft Matter, 2011, 7, 5430–5436 RSC; (c) A. Pasc, P. Gizzi, N. Dupuy, S. Parant, J. Ghanbaja and C. G-rardin, Tetrahedron Lett., 2009, 50, 6183–6186 CrossRef CAS.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed.
- (a) V. J. Nebot, J. Armengol, J. Smets, S. F. Prieto, B. Escuder and J. F. Miravet, Chem.–Eur. J., 2012, 18, 4063–4072 CrossRef CAS PubMed; (b) Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC; (c) E. Kagan, Entropy, 2010, 12, 554–569 CrossRef CAS; (d) J. M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC; (e) S. J. Rowan, S. J. Cantrill, G. R. Cousins, J. K. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef; (f) L. He, D. E. Fullenkamp, J. G. Rivera and P. B. Messersmith, Chem. Commun., 2011, 47, 7497–7499 RSC.
- (a) T. F. A. D. Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef PubMed; (b) J. D. Fox and S. J. Rowan, Macromolecules, 2009, 42, 6823–6835 CrossRef CAS.
- S. Basak, J. Nanda and A. Banerjee, Chem. Commun., 2014, 50, 2356–2359 RSC.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- (a) L. Chen, J. Raeburn, S. Sutton, D. G. Spiller, J. Williams, J. S. Sharp, P. C. Griffiths, R. K. Heenan, S. M. King, A. Paul, S. Furzeland, D. Atkins and D. J. Adams, Soft Matter, 2011, 7, 9721–9727 RSC; (b) M. A. Greenfield, J. R. Hoffman, M. O. D. L. Cruz and S. I. Stupp, Langmuir, 2010, 26, 3641–3647 CrossRef CAS PubMed.
- (a) X. Zeng, D. T. McCarthy, A. Deletic and X. Zhang, Adv. Funct. Mater., 2015, 25, 4344–4351 CrossRef CAS; (b) D. Ghosh and C. K. Das, ACS Appl. Mater. Interfaces, 2015, 7, 1122–1131 CrossRef CAS PubMed; (c) Z. Sui, X. Zhang, Y. Lei and Y. Luo, Carbon, 2011, 49, 4314–4321 CrossRef CAS.
- T. Jiao, H. Guo, Q. Zhang, Q. Peng, Y. Tang, X. Yan and B. Li, Sci. Rep., 2015, 5, 11873 CrossRef CAS PubMed.
- (a) S. Roy, A. Baral and A. Banerjee, Chem.–Eur. J., 2013, 19, 14950–14957 CrossRef CAS PubMed; (b) D. Mandal, T. Kar and P. K. Das, Chem.–Eur. J., 2014, 20, 1349–1358 CrossRef CAS PubMed.
- (a) B. Wu, S. Hou, Z. Miao, C. Zhang and J. Yanhong, Nanomaterials, 2015, 5, 1544–1555 CrossRef CAS; (b) M. Zhou, S. Liu, Y. Jiang, H. Ma, M. Shi, Q. Wang, W. Zhong, W. Liao and M. M. Q. Xing, Adv. Funct. Mater., 2015, 25, 4730–4739 CrossRef CAS.
- (a) J. G. Clar, T. Yuan, Y. Zhao, J.-C. J. Bonzongo and K. J. Ziegler, J. Phys. Chem. C, 2014, 118, 15495–15505 CrossRef CAS; (b) M. Nabeta and M. Sano, Langmuir, 2005, 21, 1706–1708 CrossRef CAS PubMed.
- Y. Yu, V. L. Pushparaj, O. Nalamasu and L. B. McGown, Molecules, 2013, 18, 15434–15447 CrossRef CAS PubMed.
- W. Li, M. Liu, H. Chen, J. Xu, Y. Gao and H. Li, Polym. Adv. Technol., 2014, 25, 233–239 CrossRef CAS.
- T. Goda, T. Kurita, T. Mitsumata and M. Sano, Chem. Lett., 2014, 43, 988–990 CrossRef CAS.
- L. Cao, M. J. Meziani, S. Sahu and Y.-P. Sun, Acc. Chem. Res., 2013, 46, 171–180 CrossRef CAS PubMed.
- (a) W. Liu, Y. Wang and Z. Li, Chem. Commun., 2014, 50, 10311–10314 RSC; (b) K. A. Heeyoung, C. Taehoon, Y. Hoon, U. Cheolsang, N. Kiju, A. Jaewook, H. Kyung and K. Lee, Carbon, 2014, 71, 229–237 CrossRef.
- (a) A. K. Das, P. P. Bose, M. G. B. Drew and A. Banerjee, Tetrahedron, 2007, 63, 7432–7442 CrossRef CAS; (b) R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC; (c) S. Basak, J. Nanda and A. Banerjee, J. Mater. Chem., 2012, 22, 11658–11664 RSC.
- (a) J. Shen, X. Xin, T. Liu, L. Tong, G. Xu and S. Yuan, J. Colloid Interface Sci., 2016, 468, 78–85 CrossRef CAS PubMed; (b) Y. Huang, M. Zeng, Z. Feng, D. Yin, Q. Xu and L. Fan, RSC Adv., 2016, 6, 3561–3570 RSC; (c) W. Cui, J. Ji, Y.-F. Cai, H. Li and R. Ran, J. Mater. Chem. A, 2015, 3, 17445–17458 RSC.
- (a) C. Hou, A. Y. Duan, Q. Zhang, H. Wang and Y. Li, J. Mater. Chem., 2012, 22, 14991–14996 RSC; (b) R. Peng, Y. Yu, S. Chen, Y. Yangc and Y. Tang, RSC Adv., 2014, 4, 35149–35155 RSC; (c) M. Zhong, Y.-T. Liu and X.-M. Xie, J. Mater. Chem. B, 2015, 3, 4001–4008 RSC.
- (a) H.-P. Cong, P. Wang and S.-H. Yu, Chem. Mater., 2013, 25, 3357–3362 CrossRef CAS; (b) J. Liu, G. Song, C. He and H. Wang, Macromol. Rapid Commun., 2013, 34, 1002–1007 CrossRef CAS PubMed; (c) D. Han and L. Yan, ACS Sustainable Chem. Eng., 2014, 2, 296–300 CrossRef CAS.
- (a) N. Kornblum and A. Scott, J. Org. Chem., 1977, 42, 399–400 CrossRef CAS; (b) P. Lan, J. J. A. Porco, M. S. South and J. J. Parlow, J. Comb. Chem., 2003, 5, 660–669 CrossRef CAS.
- D. Pan, J. Zhang, Z. Li and M. Wu, Adv. Mater., 2010, 22, 734–738 CrossRef CAS PubMed.
- (a) C. Chassenieuxa and C. Tsitsilianis, Soft Matter, 2016, 12, 1344–1359 RSC; (b) S. S. Liow, Q. Dou, D. Kai, A. A. Karim, K. Zhang, F. Xu, and X. J. Loh, ACS Biomater. Sci. Eng., 2016, 2, 295–316 Search PubMed.
- L. Lin and S. Zhang, Chem. Commun., 2012, 48, 10177–10179 RSC.
- Y. Liu and D. Y. Kim, Chem. Commun., 2015, 51, 4176–4179 RSC.
- L. Tang, Y. Wang, Y. Liu and J. Li, ACS Nano, 2011, 5, 3817–3822 CrossRef CAS PubMed.
- T. F. Jiao, F. Q. Gao, Q. R. Zhang, J. X. Zhou and F. M. Gao, Nanoscale Res. Lett., 2013, 8, 406–414 CrossRef PubMed.
- (a) S. Mallakpoura, A. Abdolmalekia and S. Borandeh, Appl. Surf. Sci., 2014, 307, 533–542 CrossRef; (b) S. Wang, Z. Lemon, I. S. Coleb and Q. Li, RSC Adv., 2015, 5, 41248–41254 RSC.
- (a) J. Lu, P. S. E. Yeo, C. K. Gan, P. Wu and K. P. Loh, Nat. Nanotechnol., 2011, 6, 247–252 CrossRef CAS PubMed; (b) X. Yan, X. Cui, B. Li and L. S. Li, Nano Lett., 2010, 10, 1869–1873 CrossRef CAS PubMed; (c) D. Pan, C. Xi, Z. Li, L. Wang, Z. Chen, B. Luc and M. Wu, J. Mater. Chem. A, 2013, 1, 3551–3555 RSC.
- Y. Hu, W. Gao, F. Wu, H. Wu, B. He and J. He, J. Mater. Chem. B, 2016, 4, 3504–3508 RSC.
- (a) Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed; (b) A. K. Das, I. Maity, H. S. Parmar, T. O. McDonald and M. Konda, Biomacromolecules, 2015, 16, 1157–1168 CrossRef CAS PubMed.
- logP was calculated by using online prediction program http://www.molinspiration.com.
- N. H. Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Wait, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef PubMed.
- C. Zhou, W. Gao, K. Yang, L. Xu, J. Ding, J. Chen, M. Liu, X. Huang, S. Wang and H. Wu, Langmuir, 2013, 29, 13568–13575 CrossRef CAS PubMed.
- V. T. Nayar, J. D. Weiland, C. S. Nelson and A. M. Hodge, J. Mech. Behav. Biomed. Mater., 2012, 7, 60–68 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEM and TEM images; NMR and mass data; FTIR and fluorescence spectra; optical image of self-healable hydrogel. See DOI: 10.1039/c6ra06587b |
| This journal is © The Royal Society of Chemistry 2016 |