
Near-white emission observed in Dy doped AlN†
Abstract
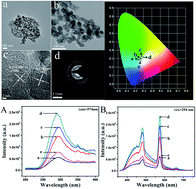
Dy doped AlN phosphors were prepared by a simple solid state route, exhibiting excellent photoluminescence. The structure, composition and morphology were investigated by means of X-ray diffraction, transmission electron microscope and energy-dispersive X-ray spectroscopy. Besides the characteristic emissions of Dy, emission from the AlN host could also be detected at room temperature. A post-thermal treatment under air atmosphere was employed to improve the photoluminescence. The blue emission and yellow emission from Dy ions contribute to the near-white emission, possessing potential applications in white LED.
 Please wait while we load your content...
Please wait while we load your content...

