
Electrocatalysis by H2–O2 membrane-free fuel cell enzymes in aqueous microenvironments confined by an ionic liquid
Abstract
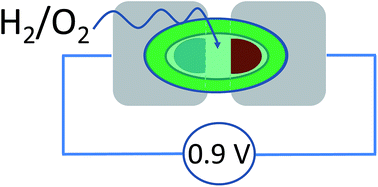
An O2-tolerant [NiFe] hydrogenase and a blue Cu oxidase exhibit excellent catalytic electrochemistry under almost dry conditions – inspiring the concept of a new type of miniature fuel cell able to provide a potential difference close to one volt. Each enzyme is immobilized on a carbon electrode that contacts an aqueous microvolume (1 μL) surrounded by an immiscible, aprotic ionic liquid. Separately, the enzymes display excellent electrocatalytic activity: brought together at a synaptic junction, an anode and cathode modified with each enzyme constitute a membrane-less fuel cell that produces over 0.8 V when equilibrated with a 96% H2–4% O2 mixture. The results show there is considerable scope for using ionic liquids to miniaturize selective enzyme fuel cells.
 Please wait while we load your content...
Please wait while we load your content...

