
An expedient synthesis of oxazolones using a cellulose supported ionic liquid phase catalyst
Abstract
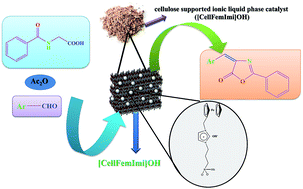
A novel cellulose supported ionic liquid phase catalyst containing hydroxide ions ([CellFemImi]OH) has been synthesized by covalent anchoring of 1-N-ferrocenylmethyl imidazole in the functionalized cellulose matrix followed by an anion metathesis reaction. The [CellFemImi]OH was characterized by various techniques including FT-IR, FT-Raman, 13C solid state NMR, X-ray diffraction, energy dispersive X-ray (EDX) analysis, field emission scanning electron microscopy (FESEM) and thermogravimetric analysis. The [CellFemImi]OH was effectively employed as a heterogeneous catalyst in the synthesis of oxazolones by cyclocondensation of aryl aldehydes with hippuric acid in the presence of acetic anhydride.
 Please wait while we load your content...
Please wait while we load your content...

