
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Radical exchange reaction of multi-spin isoindoline nitroxides followed by EPR spectroscopy†
I.
Wessely
a,
V.
Mugnaini‡
b,
A.
Bihlmeier
c,
G.
Jeschke
d,
S.
Bräse
ae and
M.
Tsotsalas
*ab
aInstitute of Organic Chemistry (IOC), Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, D-76131 Karlsruhe, Germany. E-mail: manuel.tsotsalas@kit.edu
bInstitute of Functional Interfaces (IFG), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany
cInstitute of Physical Chemistry (IPC), Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 2, D-76131 Karlsruhe, Germany
dETH Zurich, Laboratory of Physical Chemistry, Vladimir-Prelog-Weg 2, CH-8093 Zurich, Switzerland
eInstitute of Toxicology and Genetics (ITG), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany
First published on 2nd June 2016
Abstract
The synthesis of a rigid, isoindoline-functionalized tetraphenylmethane multi-spin system is described. The isoindoline nitroxide groups are used in a nitroxide exchange reaction with a TEMPO containing alkoxyamine. Using EPR spectroscopy it is possible to follow the exchange process and thereby find the optimal experimental conditions to have the maximum yield. The presented approach could be used to study the nitroxide exchange process of various systems and to determine the kinetics of the exchange process. The presented molecular components can be used as tectons in the construction of covalently linked organic networks or as model systems for EPR distance measurements.
Introduction
The synthesis of rigid organic building blocks containing multiple functional groups is of high interest to create molecular tectons for the synthesis of crystalline or amorphous covalently linked materials as well as for the assembly of non-covalently bound supramolecular architectures.1 The functionalization of these molecular tectons with nitroxide moieties lead to attractive model systems for EPR distance measurements2 and would allow the assembly of the tectons via halogen bonding3 or nitroxide exchange reaction.4 The nitroxide exchange reaction,5 which belongs to the class of dynamic covalent chemistry, has been incorporated in dynamic polymers or macromolecules6 and as a tool to trigger the self-assembly of micro-crystals.7 In addition, the nitroxide exchange reaction has been utilized to functionalize polymers, self-assembled monolayers8 or surfaces.9 Moreover, the incorporation of profluorescent nitroxides allows following the kinetics of the nitroxide exchange reaction.4 However, in all the reported cases, only the product formation could be followed, not the exchange process as such. Herein we report the synthesis of a rigid tetrahedral organic tecton containing multiple nitroxide moieties. Afterwards we performed a nitroxide exchange reaction and demonstrate that progress of the reaction can be followed by EPR spectroscopy.Results and discussion
The synthesis of the rigid multi-spin molecule is based on tetrakis(4-azidophenyl)methane (1) as a core, which is functionalized with nitroxide moieties via fourfold copper-catalysed azide alkyne click chemistry10 between the azide functions of the core 1 and the alkyne moiety of an isoindoline nitroxide 2. Scheme 1 shows the synthesis of tetraphenylmethane nitroxide (TPM-NO) 3. The TPM-NO 3 can be further functionalized using dynamic and reversible nitroxide exchange reaction. Nitroxide exchange reactions are based on thermal C–O bond homolysis of alkoxyamines, which leads to transient carbon-centred radicals and persistent nitroxide radicals. Usually these carbon centred radicals are quickly trapped by the nitroxide radicals and reform the alkoxyamines. If homolysis of an alkoxyamine is performed in presence of an additional nitroxide radical, also this additional nitroxide can trap the carbon centred nitroxide, and a mixture of the two possible nitroxides will be formed. The ratio will depend on the relative thermodynamic stabilities of the different alkoxyamines (see Fig. 1 for a schematic representation of the bond homolysis and of the nitroxide exchange reaction).11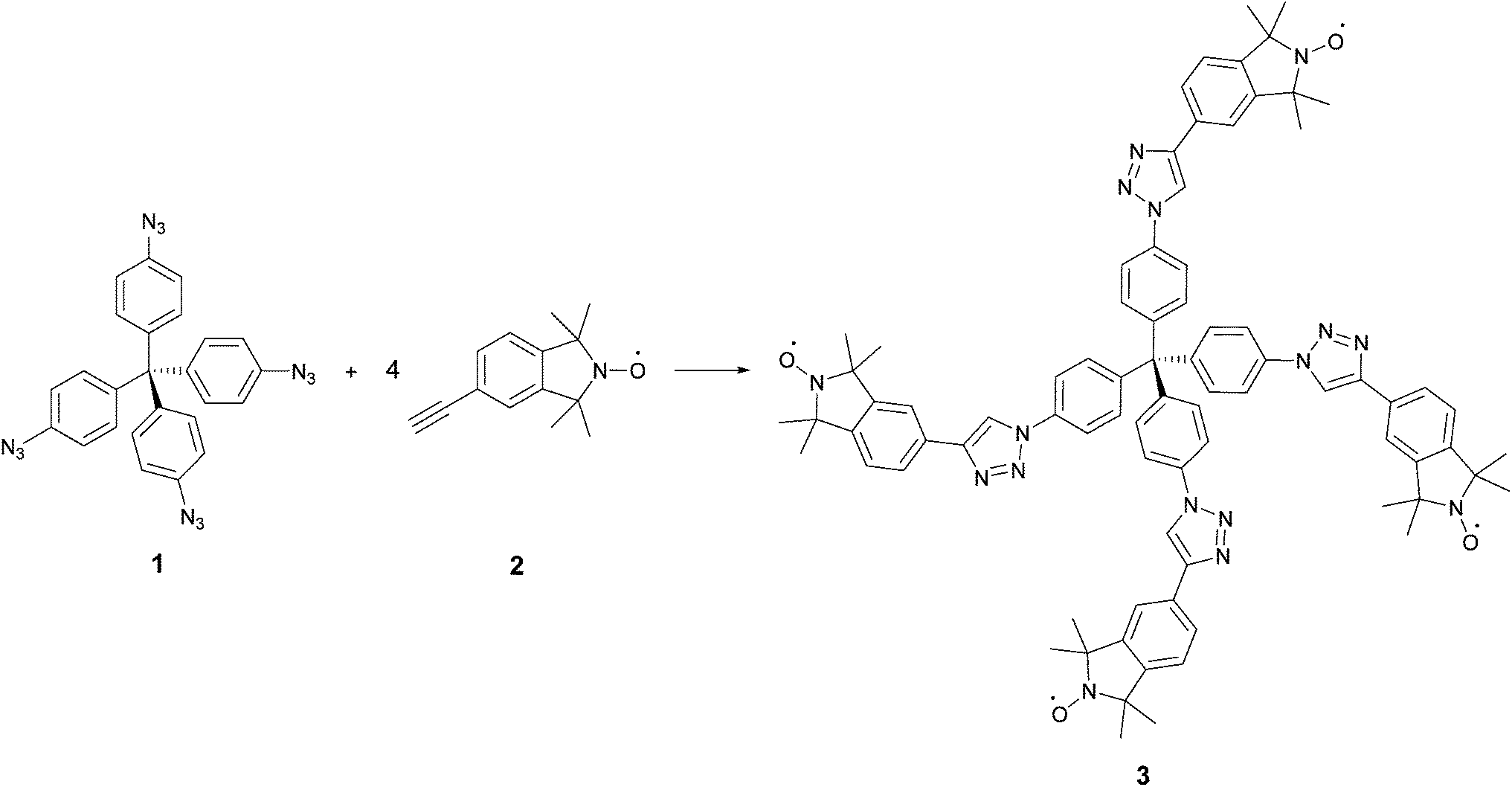 | ||
| Scheme 1 Synthesis of TPM-NO 3 using fourfold copper-catalysed azide alkyne click chemistry. | ||
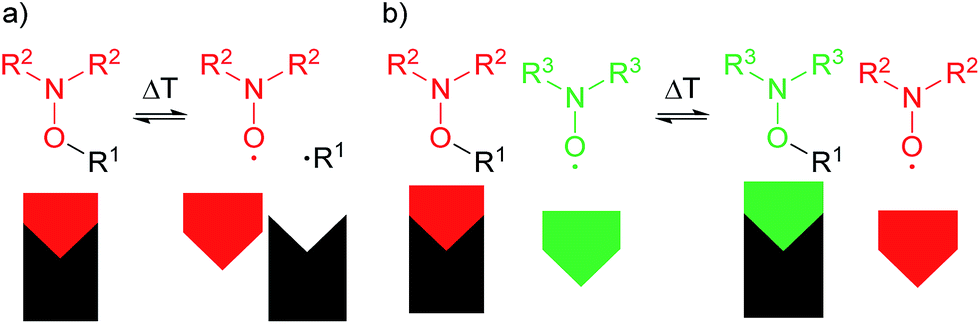 | ||
| Fig. 1 (a) Homolysis of an alkoxyamine into a nitroxide radical (red) and a carbon centred radical (black); (b) thermodynamic product formation for homolysis of an alkoxyamine in presence of an additional nitroxide radical (green). | ||
If the EPR (electron paramagnetic resonance) spectra of the two nitroxide moieties (depicted as red and green in Fig. 1) differ, i.e. in the hyperfine coupling constant as in the present case, the exchange process can be followed via EPR measurements. In order to evaluate the possibility to follow the exchange process between the isoindoline nitroxide of TPM-NO 3 and 2,2,6,6-tetramethylpiperidin-1-yloxyl (TEMPO, 5), we recorded the EPR spectra of the individual compounds and mixtures thereof in toluene at room temperature.
Fig. 2 shows the continuous wave (CW) EPR spectra of TPM-NO 3 (left) and TEMPO 5 (right) and mixtures of TPM-NO 3/TEMPO 5 with molar ratios of 9/1, 1/1 and 1/9. As can be seen from the EPR spectra in Fig. 2, the isoindoline nitroxide moiety of TPM-NO 3 has a hyperfine coupling constant of about 14.1 Gauss. The EPR spectra show the typical tumbling induced line profile. The TEMPO nitroxide 5 instead has a hyperfine coupling constant of about 15.5 Gauss. Due to the difference in the hyperfine coupling constants of the isoindoline and TEMPO nitroxides, the exchange reaction can be easily followed by means of EPR.
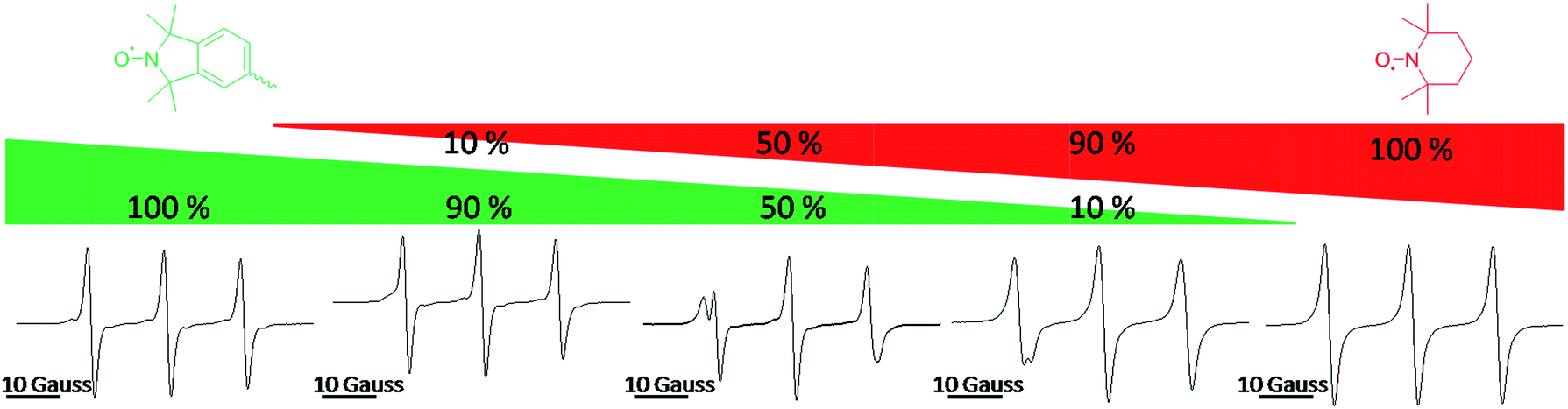 | ||
| Fig. 2 Continuous wave (CW) EPR spectra of TPM-NO 3 (left) and TEMPO 5 (right) and mixtures of TPM-NO 3/TEMPO 5 with molar ratios 9/1, 1/1 and 1/9. | ||
We performed the nitroxide exchange reaction of the multi-spin system TPM-NO 3 with the TEMPO-alkoxyamine 4 shown in Scheme 2. Firstly, we followed the exchange reaction between TPM-NO 3 and TEMPO-alkoxyamine 4 using an equimolar ratio (with respect to the nitroxide moieties). Compounds 3 and 4 were dissolved in toluene and mixed at room temperature. The resulting solution was divided in several aliquots, inserted in closed ampules and degassed via bubbling with argon. Afterwards the different aliquots were heated under argon at 80 °C for either 15 min, 1 h, 2 h, 14 h, 48 h or 96 h. After cooling to room temperature the continuous wave EPR spectra were recorded. Fig. 3 (top) shows the CW EPR spectra for the different points in time.
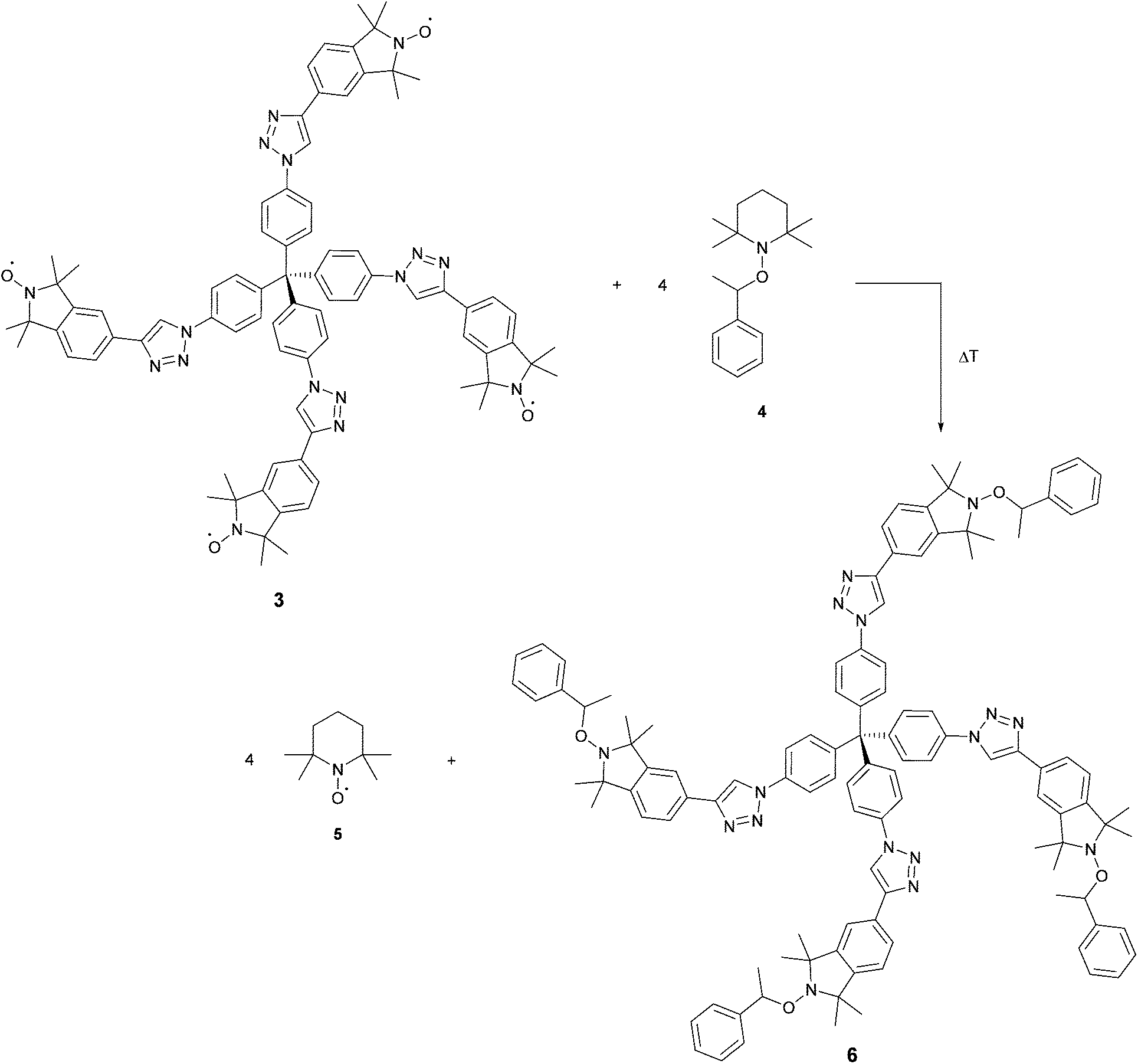 | ||
| Scheme 2 Nitroxide exchange reactions of TPM-NO 3 and TEMPO-alkoxyamine 4. | ||
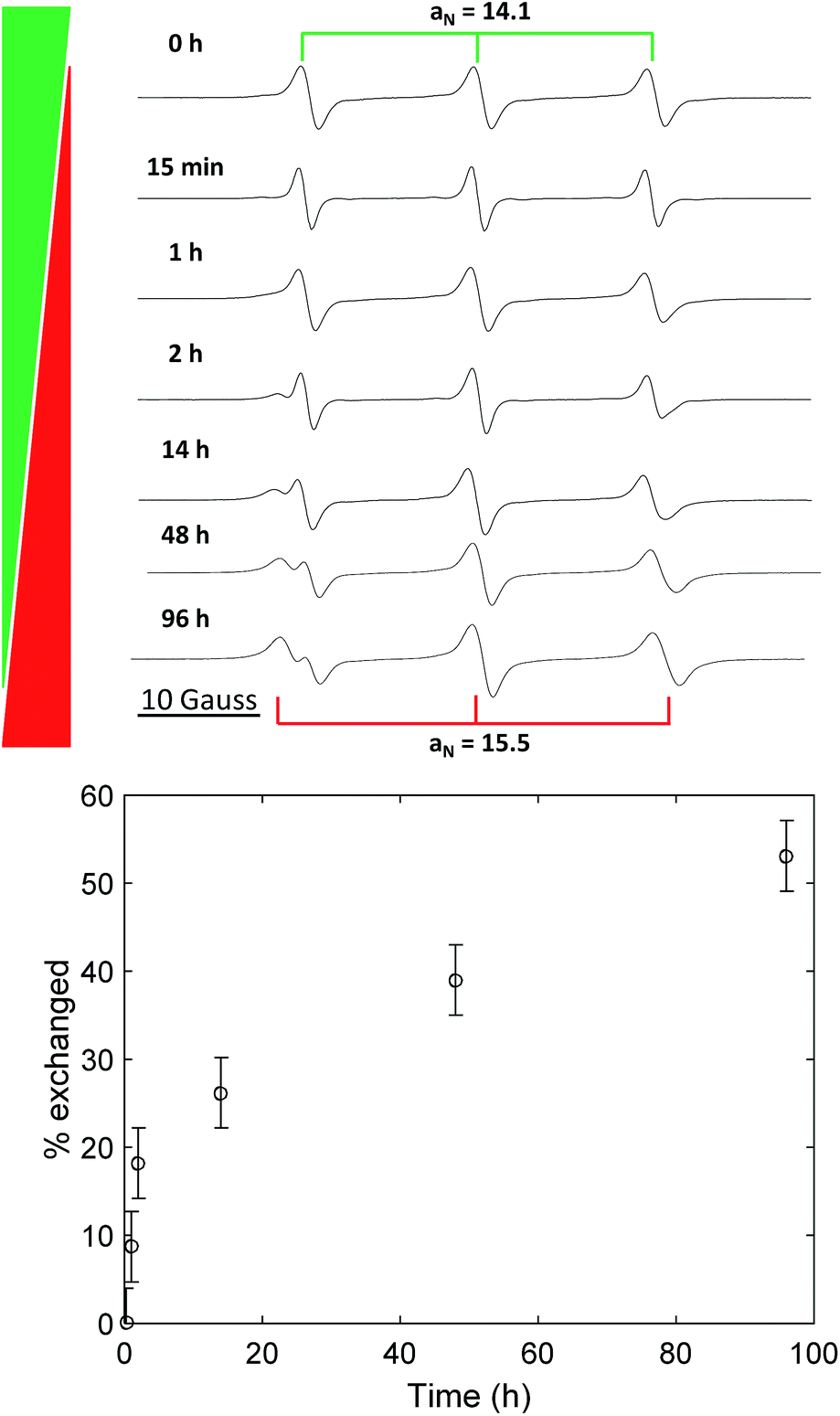 | ||
| Fig. 3 Top: EPR spectra of TPM-NO 3/TEMPO-alkoxyamine 4 1/1 mixture in toluene without heating (0 °C) and after heating at 80 °C for 15 min, 1 h, 2 h, 14 h, 48 h and 96 h. Bottom: graph showing the percentage of exchanged nitroxides versus time. | ||
In the EPR spectra of Fig. 3 (top), one can clearly see the progress of the exchange reaction over time, with a decrease in relative intensity of the species with lower hyperfine coupling constant (TPM-NO 3) and an increase of TEMPO 5, showing a larger hyperfine coupling constant. From the fitting of the spectra (for details see the ESI†), we could obtain the different relative percentages of the two species and could plot the exchange percentage as function of time (see Fig. 3, bottom). The plot reveals that the exchange process is a fast initial reaction (within the first two hours) followed by a deceleration. The deceleration is most likely due to the decreasing concentration of TEMPO-alkoxyamine 4 in combination with the competition for radical trapping between an increasing concentration of liberated TEMPO 5 and decreasing concentration of TPM-NO 3.
Even after 96 h, the exchange reaction does not seem to have reached the thermodynamic limit, where both species adapt their final equilibrium concentrations. In order to find conditions under which the equilibrium is reached faster and in order to optimize the yield of TPM-NO-alkoxyamine 6, we investigated the influence of both the reaction temperature and the equivalents of TEMPO-alkoxyamine 4 on the exchange process. Fig. 4 shows the comparison of the CW EPR spectra of TPM-NO 3 and TEMPO-alkoxyamine 4 in mixtures of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (a), 1:2 (b), and 1:5 (c) molar ratios (related to nitroxide moieties), after heating at 100 °C for 1 h and 24 h.
:1 (a), 1:2 (b), and 1:5 (c) molar ratios (related to nitroxide moieties), after heating at 100 °C for 1 h and 24 h.
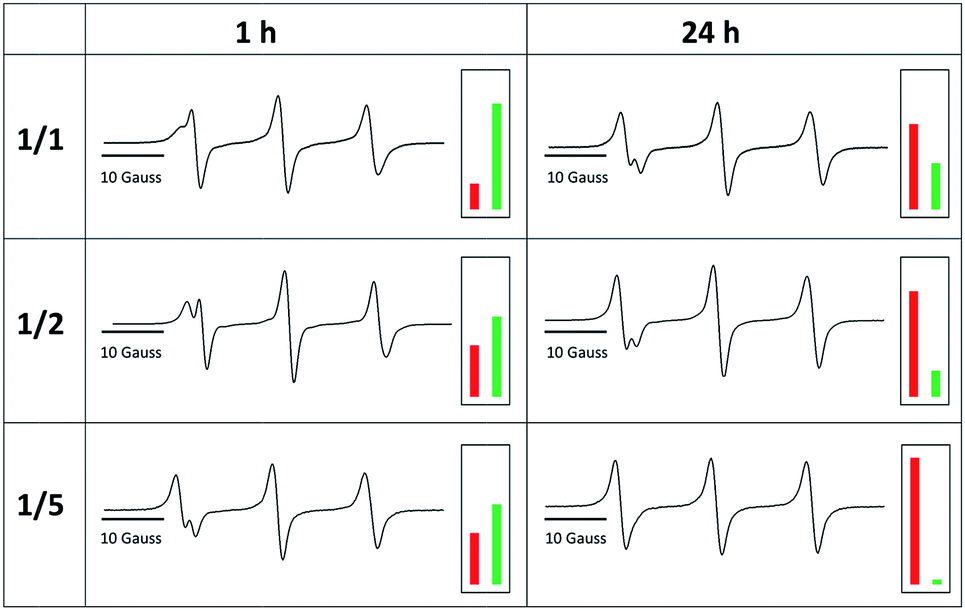 | ||
| Fig. 4 CW EPR spectra of TPM-NO 3 and TEMPO-alkoxyamine 4 after heating at 100 °C for 1 h and 24 h. Top: 1/1 molar ratio of TPM-NO 3/TEMPO-alkoxyamine 4; middle: 1/2 molar ratio of TPM-NO 3/TEMPO-alkoxyamine 4; bottom: 1/5 molar ratio of TPM-NO 3/TEMPO-alkoxyamine 4. | ||
The EPR spectra show that the exchange reaction is almost quantitative after 24 h at 100 °C in the presence of 5 equivalents of TEMPO-alkoxyamine 4 with 96% yield (and 77% yield after 1 h), whereas the 1/1 (20% yield after 1 h and 65% after 24 h) and 1/2 mixtures (39% yield after 1 h and 80% yield after 24 h) still contain large amounts of isoindoline nitroxide moieties of TPM-NO 3.
To verify if the exchange reaction reaches equilibrium at 100 °C after 24 h, we determined the expected thermodynamic limit of exchange for the investigated molar ratios. For this, we first estimated the Gibbs free energy ΔG by performing quantum chemical calculations. Employing density functional theory methods as implemented in the TURBOMOLE program package,12 we obtain values between −4 kJ mol−1 and +5 kJ mol−1. Considering the error in the calculated ΔG, we evaluated the limit of exchange for several values in the range of ±15 kJ mol−1 (for details of the calculations see Section 5 in the ESI†). The experimental results for T = 100 °C after 24 h and the calculated results for selected values of ΔG are summarized in Table 1. The calculated results for slightly negative values of ΔG fit well to the experimentally observed data. The calculations suggest that the exchange process is approaching its thermodynamic limit of exchange after heating at 100 °C for 24 h and that a nearly quantitative exchange can be reached if the TEMPO-alkoxyamine 4 is present in excess.
| Molar ratio of 3/4 | Calculated for ΔG = −10 kJ mol−1 | Calculated for ΔG = −5 kJ mol−1 | Calculated for ΔG = 0 kJ mol−1 | Experimental after 24 h at T = 100 °C |
|---|---|---|---|---|
| 1/1 | 66% | 58% | 50% | 65% |
| 1/2 | 97% | 91% | 82% | 80% |
| 1/5 | 100% | 100% | 100% | 96% |
Experimental
The EPR spectra in this work were recorded on a Bruker ESP300E spectrometer. The compounds were dissolved in toluene and deoxygenated by bubbling argon for several minutes. After treatment of the samples as written in the text the spectra were taken at 298 K.The instrument settings were as follows: microwave power 2.00 mW, modulation amplitude 0.0452 mT, modulation frequency 100 kHz, scan time 180 s. Further details as well as the synthesis of the compounds are described in the ESI.†
Conclusions
Here we describe the successful synthesis of novel rigid isoindoline based multispin nitroxides via fourfold click reaction. These nitroxides were further converted into alkoxyamines via exchange reaction with a TEMPO-alkoxyamine and characterized using EPR spectroscopy. Our results demonstrate that EPR spectroscopy is a versatile tool to follow the exchange process and to determine several factors influencing the kinetics of the exchange process and optimize the experimental conditions to have the maximum yield. The presented approach could be used to study the nitroxide exchange process of various systems and the presented molecular components can be used as initiators for nitroxide mediated polymerization (NMP)13 or as tectons in the construction of supramolecular and covalently linked organic networks. Further investigations in this regards are currently ongoing.Acknowledgements
We acknowledge the SFB 1176 funded by the German Research Council (DFG) in the context of projects C5 (M. Tsotsalas) and Q4 (A. Bihlmeier) as well as the Studienstiftung and the Carl-Zeiss-Stiftung for funding. We thank Dr Laure Monnereau for authentic samples of 1 and Dr Fairful-Smith for authentic samples of 2.Notes and references
- T. Muller and S. Bräse, RSC Adv., 2014, 4, 6886–6907 RSC.
- S. Valera, J. E. Taylor, D. S. B. Daniels, D. M. Dawson, K. S. Athukorala Arachchige, S. E. Ashbrook, A. M. Z. Slawin and B. E. Bode, J. Org. Chem., 2014, 79, 8313–8323 CrossRef CAS PubMed.
- G. R. Hanson, P. Jensen, J. McMurtrie, L. Rintoul and A. S. Micallef, Chem.–Eur. J., 2009, 15, 4156–4164 CrossRef CAS PubMed.
- J. P. Blinco, K. E. Fairfull-Smith, A. S. Micallef and S. E. Bottle, Polym. Chem., 2010, 1, 1009–1012 RSC.
- C. J. Hawker, G. G. Barclay and J. Dao, J. Am. Chem. Soc., 1996, 118, 11467–11471 CrossRef CAS.
- H. Otsuka, Polym. J., 2013, 45, 879–891 CrossRef CAS.
- B. Schulte, M. Tsotsalas, M. Becker, A. Studer and L. De Cola, Angew. Chem., Int. Ed., 2010, 49, 6881–6884 CrossRef CAS PubMed.
- H. Wagner, M. K. Brinks, M. Hirtz, A. Schäfer, L. Chi and A. Studer, Chem.–Eur. J., 2011, 17, 9107–9112 CrossRef CAS PubMed.
- M. Becker, L. D. Cola and A. Studer, Chem. Commun., 2011, 47, 3392–3394 RSC.
- O. Plietzsch, C. I. Schilling, M. Tolev, M. Nieger, C. Richert, T. Muller and S. Brase, Org. Biomol. Chem., 2009, 7, 4734–4743 CAS.
- H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS PubMed.
- V. TURBOMOLE, Program Package for ab initio Electronic Structure Calculations, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH 1989–2007, TURBOMOLE GmbH, 2007, http://www.turbomole.com Search PubMed.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, synthetic procedures, details on EPR measurements/fits. See DOI: 10.1039/c6ra06510d |
| ‡ Current address: International Iberian Nanotechnology Laboratory, Avenida Mestre José Veiga, 4715-330, Braga, Portugal. |
| This journal is © The Royal Society of Chemistry 2016 |