
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMelt processability and thermomechanical properties of blends based on polyhydroxyalkanoates and poly(butylene adipate-co-terephthalate)†
Matilda
Larsson
ab,
Olivia
Markbo
a and
Patric
Jannasch
*a
aPolymer & Materials Chemistry, Department of Chemistry, Lund University, P.O. Box 124, SE-221 00, Lund, Sweden. E-mail: patric.jannasch@chem.lu.se
bBaxter Healthcare, Material and Platform, Magistratsvägen 16, SE-224 41, Lund, Sweden
First published on 28th April 2016
Abstract
The limited thermal stability of polyhydroxyalkanoates (PHAs) hinders their wide applicability, and methods to improve the processability of these biopolyesters are needed for efficient processing, e.g. by melt extrusion. In the present study we have shown by isothermal gravimetry, dynamic rheology and molecular weight analysis that the thermal stability of the PHAs at the processing temperature can be dramatically improved by simply washing the materials in a 1 mM aqueous HCl solution. Hence, the thermal decomposition temperature increased by up to 50 °C after the treatment. Subsequently, treated poly(3-hydroxybutyrate) and poly(3-hydroxybutyrate-co-4-hydroxybutyrate) were blended with different amounts of poly(butylene adipate-co-terephthalate) by melt extrusion in order to further enhance the processability and thermomechanical properties. Microscopy of freeze fractured samples of the biodegradable blends showed phase separated blends with poor interfacial adhesion. Melt rheology and dynamic mechanical analysis results indicated a phase inversion between 60 and 80 wt% of the respective PHA. After adding dicumyl peroxide during the extrusion, the interfacial adhesion improved significantly, and the dynamic shear and tensile storage modulii increased with increasing content of the peroxide. The results of the present study demonstrate that an acid wash may significantly improve processability of PHAs, and that combinations of blending and reactive extrusion can be employed to further enhance and tune the thermomechanical properties of the materials.
Introduction
Polyhydroxyalkanoates (PHAs) are biobased and biodegradable polyesters that are accumulated as intracellular granules by many naturally occurring bacteria and are potential alternatives to plastics made from fossil sources. The most extensively studied PHA is poly(3-hydroxybutyrate) [P(3HB)], which is a highly crystalline and brittle polymer with limited processability.1–3 The inherently poor thermal stability of P(3HB) is a major concern and thermal decomposition by chain scission starts at temperatures close to the melting point, making processing in the melt state a considerable challenge.4 One strategy to improve the processability of PHAs is to employ copolymers such as poly(3-hydroxybutyrate-co-hydroxyvalerate) (PHBV) and poly(3-hydroxybutyrate-co-4-hydroxybutyrate) [P(3,4HB)]. The melting point of PHBV is lower than P(3HB) and the processing of P(3HB) and PHBV, respectively, has been studied on the laboratory scale by extrusion, as well as injection and compression molding.5–10 P(3,4HB) has lower crystallinity and melting point than P(3HB) which broadens the processing window.11–13Another approach to improve the properties of PHAs is to prepare blends with another biodegradable polymer.14 PHAs have previously been melt blended with biodegradable polymers such as poly(lactic acid) (PLA),14–18 poly(butylene succinate) (PBS),19 poly(ε-caprolactone) (PCL),20 lignin,21 poly(butylene adipate-co-terephthalate) (PBAT),22,23 and PBAT/starch.24 PBAT is a synthetically derived biodegradable polymer, suitable for film extrusion. It has a high elongation at break, is ductile and has potential to toughen the brittle P(3HB).22 Javadi et al. have previously studied injection molding of PHBV/PBAT blends.22 They found the specific toughness (with density reduction taken into account) and the strain at break to increase, but the specific modulus and strength decreased with increasing content of PBAT. In addition, the crystallinity of PHBV decreased with increasing PBAT content. PBAT has previously been blended with PLA to increase the tensile toughness of the material and PBAT also acted as a lubricant during the extrusion.25 In addition, PBAT has been blended with thermoplastic starch26,27 and PBS.28
A further factor of high relevance for the processability is the degree of purity achieved after the biosynthesis and extraction of the PHAs. The purity of the polymers affects both the thermal stability during processing and the resulting mechanical properties.29 Up until now there are only a few reports in the literature that consider the influence of the purity on the thermal stability of PHA before processing.30,31
The primary aim of the present study was to explore possibilities to improve the processability of PHA materials by blending with PBAT, and to further enhance and tune the properties by reactive extrusion. PBAT was selected because of its good processability and the possibility to obtain a soft matrix for the considerably more brittle PHAs. Dicumyl peroxide (DCP) has previously been used in reactive processing of P(3HB)/PBS and P(3HB)/PLA blends, which resulted in an increased compatibility between the components of the respective polymer blend.19,32 In the present case, the properties and morphology of the biodegradable blends were evaluated by rheometry, dynamic mechanical analysis, calorimetry, gravimetry and microscopy. As a first important step, the possibilities to increase the thermal stability of the PHA materials were investigated by employing different purification strategies. The effect was studied by means of thermal gravimetry, rheometry and molecular weight measurements.
Experimental
Materials
P(3HB) powder (ENMAT Y3000P) was purchased from Tianan Biologic Material Co. Ltd., Ningbo, China, and P(3,4HB) powder (SoGreen 00A-1) was purchased from Tianjin Green Bioscience, China. The fraction of 4HB units in the copolymer was 3 mol% as determined by 1H NMR spectroscopy. Granules of PBAT Ecoflex F Blend C1200 (LOT: 00522875L0) were kindly provided by BASF (Germany). DCP, (Sigma-Aldrich, 98% purity), chloroform (Fisher Chemical, analytical reagent grade), methanol (Scharlau, ASC basic) and HCl (Honeywell, 37% aq. solution) were used as received. PHA was pre-treated as described below. Both PHA and PBAT were dried at 50 °C under vacuum over night before extrusion.Polymer pre-treatment
Two different PHA pre-treatment methods were evaluated, namely soxhlet extraction with CHCl3 and washing in a 1 mM aqueous HCl solution. In the first procedure, the extraction was conducted during 5 h at 90 °C where the PHA was successively dissolved in CHCl3. PHA was then precipitated in methanol and subsequently filtered and dried during 24 h at 50 °C under vacuum. In the second treatment method, the washing was performed during 30 min at room temperature. Next, the PHA was washed 3 times with deionized water, filtered and dried for 24 h at 50 °C under vacuum.30 The aq. HCl wash was chosen as the purification method for PHA in the following studies.Preparation of blends
A series of blends (15 g) with weight ratios of P(3HB)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PBAT equal to 100:0, 80:20, 60:40, 40:60, 20:80, 0:100 were prepared in a 15 cm3 DSM midi twin screw extruder. The melt extrusion was conducted with a temperature profile from feeder to die of 150–175–182 °C and at a rotational rate of 50 rpm, keeping an average residence time of 1 min. Blends of P(3,4HB) and PBAT were prepared in weight ratios equal to 100:0, 80:20, 60:40, 50:50, 40:60, 20:80 and 0:100 at a rotational rate of 50 rpm with an average residence time of 2 min. The temperature profile from feeder to die was 140–150–160 °C. In all the blending procedures, the two polymer components were added to the extruder all at once, and the average residence time was determined from the time all polymers had been fed to the extruder up until the outlet was opened. DCP was added at the end of the feeding together with the dry mixed polymers.
:PBAT equal to 100:0, 80:20, 60:40, 40:60, 20:80, 0:100 were prepared in a 15 cm3 DSM midi twin screw extruder. The melt extrusion was conducted with a temperature profile from feeder to die of 150–175–182 °C and at a rotational rate of 50 rpm, keeping an average residence time of 1 min. Blends of P(3,4HB) and PBAT were prepared in weight ratios equal to 100:0, 80:20, 60:40, 50:50, 40:60, 20:80 and 0:100 at a rotational rate of 50 rpm with an average residence time of 2 min. The temperature profile from feeder to die was 140–150–160 °C. In all the blending procedures, the two polymer components were added to the extruder all at once, and the average residence time was determined from the time all polymers had been fed to the extruder up until the outlet was opened. DCP was added at the end of the feeding together with the dry mixed polymers.
Characterization methods
The average molecular weights of the PHAs and PBAT were determined by size exclusion chromatography (SEC). Three Shodex columns (KF-805, KF-804, KF-802.5) connected to a Viskotek refractometer/viscometer 250, was used with CHCl3 as solvent for measurements performed at room temperature at a solvent flow rate of 3 ml min−1. A calibration curve was constructed using polystyrenes (Mw = 650 kDa, Water Associates; 96 kDa, Polymer Laboratories; 30 kDa, Polyscience Inc.; and 3.18 kDa, Agilent Technologies) as standards.Scanning Electron Microscopy (SEM JEOL JSM-6700F) was used to characterize the blend morphology of freeze fracture surfaces of the blends. The samples were fractured in liquid nitrogen and sputtered-coated with a 10 nm layer of Au–Pd. The microscope was operated at an accelerating voltage of 15 kV. In addition, the cross-linked samples were fractured at room temperature at a strain rate of 5 mm min−1.
Thermogravimetric analysis (TGA) was performed on a TA Instruments TGA Q500. Samples of 3–5 mg were heated to 550 °C at a heating rate of 10 °C min−1 under nitrogen. Decomposition temperatures (Td) were determined at the maximum weight loss rate of the samples. The weight fraction of PHA was determined as the percentage change in weight loss between 200 and 310 °C. Isothermal measurements to study the decomposition of P(3HB) and P(3,4HB) under air were performed at 180 and 160 °C, respectively, during 8 h. Differential scanning calorimetry (DSC) measurements were carried out on a TA Instruments DSC Q2000. Samples were heated to 185 °C at a heating rate of 10 °C min−1, thereafter cooled to −70 °C and kept isothermal for 3 min before heated again to 200 °C at the same heating rate. The crystallinity of the PHB phase was calculated using the equation Xc (%) = (ΔHf,PHB/ΔH0PHB) × (100/W), where W is the weight fraction of PHB in the blend, measured by TGA, and ΔH0 is the heat of fusion of 100% PHB (146 J g−1).33 The heat of fusion for the PHB phase in the blends was determined from the second heating scan.
Dynamic mechanical analysis (DMA) was performed on a TA Instruments DMA Q800. Specimens from the extruded material were hot-pressed into rectangular shapes of approximately 15 × 9 × 1 mm3 using a hydraulic press (Specac, GS15011) at 160 °C [P(3,4HB)] or 180 °C [P(3HB)] during 2 min, and subsequently cooled to room temperature between two metal blocks. The samples were analyzed at 1 Hz in the temperature interval −60 to 100 °C at a heating rate of 3 °C min−1. The measurements were performed in the linear viscoelastic region at a strain of 0.05%. The glass transition temperature (Tg) was determined as the position of the maximum peak value of the loss modulus. Dynamic rheology measurements were carried out using a TA Instruments Advanced Rheometer AR2000 ETC. Measurements were performed using parallel plates (∅ = 15 mm) during 40 min at 170 and 180 °C, respectively, and 1 Hz under nitrogen atmosphere. A 2% strain was used which was within the linear viscoelastic region. The specimens (∅ = 15 mm, h = 1 mm) were hot-pressed from the extruded parts as described above.
Results and discussion
Pre-treatment and stabilization of the PHAs
The practical processing temperatures of PHAs are essentially set by their crystalline melting points (Tm) because of their poor thermal stability in the melt state. The initial thermal decomposition temperatures of the as-received P(3HB) and P(3,4HB) were Td = 270 and 240 °C, respectively, under nitrogen. We were initially interested to investigate how much the value of Td could be raised by a straightforward pre-treatment, and especially to clarify the effects of such treatments on the thermal stability at the melt processing temperature. A stability study was performed on both the PHAs after soxhlet extraction with CHCl3 and after washing with aq. HCl solution, respectively. Arza et al. recently reported on enhanced thermal stability after an aqueous acid treatment before solution casting of PHB.30 Following this work, Lopez-Abelairas and co-workers demonstrated that acid treatment in the recovery of PHA from bacteria yielded samples with high purity and low levels of degradation, as compared to other methods.31PHAs generally have good solubility in CHCl3. However, during the soxhlet extraction an insoluble fraction remained in the thimble. Unfortunately, this fraction prevented the filtration necessary to study the dissolved polymer fraction by, e.g., NMR spectroscopy. After washing with aq. HCl and soxhlet extraction with CHCl3, respectively, the Td value of P(3,4HB) increased by up to 50 °C under nitrogen, as determined by TGA (Fig. 1a, and ESI Fig. S1† for corresponding data for P(3HB)). The increase in Td represented a significant increase in the thermal stability. Considering the isothermal TGA data at the intended processing temperatures of P(3HB) (180 °C) and P(3,4HB) (160 °C), the positive effect of the acid treatment was quite remarkable (Fig. 1b). As seen, the as-received P(3,4HB) decomposed much faster in comparison to the acid-treated sample. After 1 h at 160 °C under air, the weight loss of the as-received sample was 10% while the weight loss of the acid-treated sample was below 1%. The corresponding weight losses after 8 h were 95 and approx. 1 wt%, respectively. For P(3HB) the initial weight loss after 1 h was below 1 wt% for both the as-received and acid-treated samples. However, after 8 h the difference between the samples was significant, 99 wt% loss of the as-received P(3HB) compared to less than 10 wt% of the acid-treated sample. Hence, the thermal stabilities of both materials were significantly higher after pre-treatment.
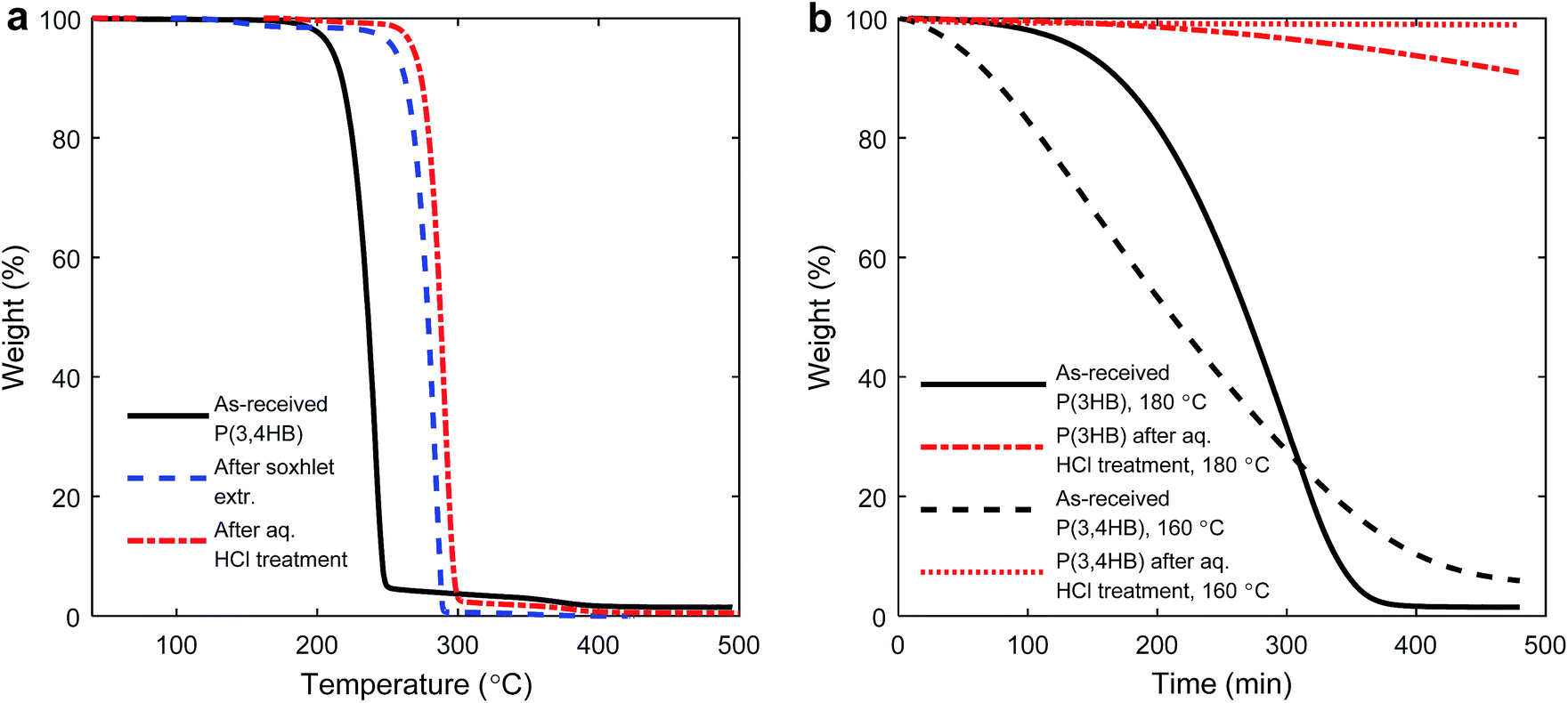 | ||
| Fig. 1 TGA traces of as-received and pre-treated P(3,4HB) recorded at 10 °C min−1 under nitrogen (a) and TGA traces showing the isothermal decomposition of P(3,4HB) and P(3HB) materials under air during 8 h at 160 and 180 °C, respectively (b). | ||
As seen in Table 1, both Tm and Tc increased after the pre-treatments of the polymers. However, the increase in Tm was approximately 10 °C, compared to the 50 °C increase of Td due to the pre-treatment. Hence, the processing window was favored by both treatment strategies. The simultaneous increase in Td, Tm and Tc may indicate higher purity of the polymer after pre-treatment. In contrast, the crystallinity (Xc) of the P(3,4HB) was not affected by any of the treatment methods, but increased with the pre-treatment of P(3HB).
| Sample | T d (°C) | T m (°C) | T c (°C) | T g (°C) | X c (%) | G′b (kPa) |
|---|---|---|---|---|---|---|
| a Measured by TGA and DSC (ESI, Fig. S1 and S2). b G′ after 5 minutes at 180 °C for P(3HB) and 170 °C for P(3,4HB) as measured by rheology. | ||||||
| P(3HB) | ||||||
| As-received | 270 | 180 | 132 | 4 | 53 | 35 |
| Soxhlet extr. | 288 | 183 | 133 | 7 | 63 | 38 |
| Acid wash | 290 | 182 | 133 | 5 | 72 | 35 |
|
||||||
| P(3,4HB) | ||||||
| As-received | 240 | 158 | 86 | −1 | 49 | 0.67 |
| Soxhlet extr. | 280 | 169 | 100 | 4 | 47 | 17 |
| Acid wash | 290 | 170 | 107 | 0 | 48 | 140 |
The positive effect of the pre-treatment was also demonstrated by dynamic rheology measurements. As seen in Fig. 2a, the shear storage modulus was significantly higher for the pre-treated polymers. The best effect was seen for the acid-treated P(3,4HB) which had a higher initial modulus and a very moderate loss in modulus, indicating a far less reduction in molecular weight. The modulus for the as-received and soxhlet extracted polymer decreased by approximately two decades over 40 min, respectively, due to thermal degradation that occurred at 170 °C. The rheological properties can be related to the molecular weight, and a reduction in modulus to polymer chain scission reactions.34 The effect of the pre-treatments on the melt shear storage modulus for P(3HB) was not obvious from the rheology measurements (ESI, Fig. S3a†). Still there was a significant increase in Td (10 °C) for the samples after both of the pre-treatment methods (ESI, Fig. S2†). From the TGA and rheology measurements it was clear that the most efficient pre-treatment method was washing with a diluted HCl solution. Although Tm increased with 10 °C after acidic treatment of P(3,4HB) the increase in Td was even higher. This should increase the processing temperature window of the PHAs, making the acid-wash the most efficient pre-treatment method also for this polymer.
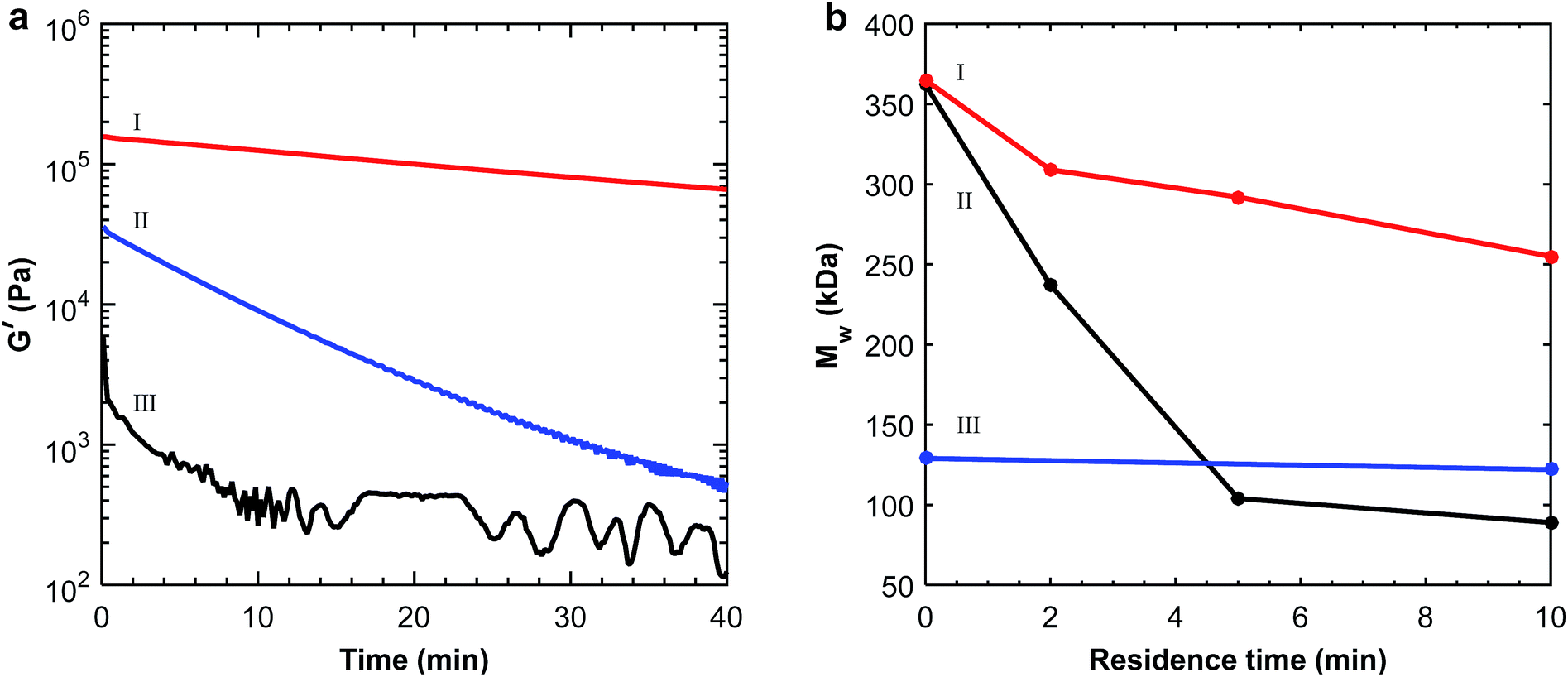 | ||
| Fig. 2 Degradation observed by (a) monitoring the melt shear storage modulus of P(3,4HB): (I) treated with aq. HCl solution, (II) soxhlet extracted with CHCl3 and (III) as-received, and by (b) following the molecular weight measured by SEC of (I) HCl-treated P(3,4HB), (II) as-received P(3,4HB) and (III) PBAT. | ||
The stability of the pre-treated polymers and the as-received PHAs was also assessed by molecular weight measurements after different residence times in the extruder. As seen in Fig. 2b for P(3,4HB), the molecular weight before extrusion was the same for the as-received and the acid-treated samples, indicating no significant chain degradation effects due to the acid treatment. The non-treated polymer degraded extensively during processing for 10 min, while the acid-treated polymer retained a much higher molecular weight. The largest decrease in molecular weight for the non-treated P(3,4HB) was seen during the first 5 min, during which the molecular weight decreased by 71%, compared to only 20% for the acid-treated polymer, making the former polymer very sensitive to the residence time in the extruder. Before processing, it was not possible to measure the molecular weight of the as-received P(3HB) by SEC, due to solubility difficulties. However, after 2 min of processing it was possible to compare the non-treated P(3HB) with the acid-treated P(3HB) (ESI, Fig. S3b†). SEC measurements of the non-treated P(3HB) and acid-treated P(3HB) after 2 min of processing revealed a difference in molecular weight of 17%. Notably, there was no reduction in molecular weight for PBAT after extrusion at 170 °C (Fig. 2b).
In conclusion, it is essential to assess and improve the thermal stability of PHAs prior to melt processing. Obviously, the thermal stability can be significantly improved at the processing temperature by a proper pre-treatment. Csomorova et al. have reported on metal ions that seem to accelerate the thermal degradation of PHB,35 but the information on this issue is still limited. Because of the biobased nature of PHAs there are likely to be batch-to-batch variations during production. By employing an additional pre-treatment the thermal stability can be significantly enhanced while concurrently minimizing these variations. In addition to the improved thermal and processing properties, the acid-treatment has a lower environmental impact and is more cost effective compared to the use of halogenated solvents such as CHCl3.29
PHA–PBAT blends
The effect of blending P(3HB) and P(3,4HB), respectively, with PBAT on the processing properties, the morphology and the physical properties were investigated. PBAT is a synthetic biodegradable polymer especially suited for film extrusion. It is a soft polymer with Td = 393 °C and Tm = 140 °C, in contrast to P(3HB) and P(3,4HB) which are brittle materials with a high crystallinity. By blending P(3HB) and P(3,4HB), respectively, with PBAT, the goal was to reduce brittleness and improve processability.Since PHA is sensitive to high temperatures and shear forces, it is important to carefully set the process parameters. The processing temperature must be high enough for the polymers to melt, but as low as possible to minimize thermal decomposition. In order to obtain adequate mixing the residence time needs to be sufficient, but still as short as possible to avoid thermal decomposition. The rotational rate and shear forces are also important where a high rate reduces the processing time, but a too high rate will lead to increased decomposition. Previously it has been reported that the molecular weight of P(3HB) and PHBV decreased with increasing processing temperature and residence time,7 and that the shear forces significantly contribute to the molecular weight reduction.5 In the present study, 50 rpm was chosen because a higher rotational rate caused a too low viscosity of the melt. An average residence time of 1 to 2 min was sufficient to obtain proper mixing while also minimizing the decline in molecular weight. A temperature gradient along the barrels was chosen to reduce the residence time at the highest temperatures. The two polymer components of the blend were added simultaneously, PBAT in granulate form and the PHAs in the form of pellets from pressed powder. Notably, only acid-treated PHAs were used in the blending study. The thermal degradation of the as-received polymers was too extensive for the extrusion, resulting in highly discoloured melts with very low viscosities.
:PBAT blends. Phase separation was clearly visible in the blends with weight ratios of P(3HB):PBAT equal to 20:80 (Fig. 3a) and 80:20 (Fig. 3d) due to the rather poor interfacial adhesion between the phases. The phase separation was less visible for the blends with the 40:60 (Fig. 3b) and 60:40 (Fig. 3c) compositions. For the sample with 20:80 composition, PBAT was the continuous phase with P(3HB) domain sizes of approx. 1–2 µm. A phase inversion seemingly occurred for the 80:20 sample where P(3HB) was the continuous phase with PBAT domain sizes of approx. 1 µm. The phase separation of the P(3,4HB):PBAT blends was not as visible as for the P(3HB):PBAT blends, which may be due to a higher degree of compatibility of the components at the interface or efficient mechanical interlocking of the phases (ESI, Fig. S5†).
 | ||
| Fig. 3 SEM images of cryo fractured surfaces of P(3HB):PBAT blends with (a) 20:80, (b) 40:60, (c) 60:40 and (d) 80:20 weight ratios. | ||
:20 blend composition. The Tg of P(3HB) was difficult to detect by DSC due to the highly crystalline nature of the polymer, and the value varied between 2 and 9 °C without any obvious trend. The crystallinity of P(3HB) and P(3,4HB) in the blends is given in Fig. 4. There was a significant difference in crystallinity between the samples containing 20 and 40 wt% P(3HB) but the value was relatively constant at higher P(3HB) contents. As seen, there was a slight decrease in crystalline content of P(3,4HB) between the samples containing 40 and 100 wt% P(3,4HB). A previous study of PLA/PBAT blends showed that the crystallinity of PLA was not increased by the blending with PBAT up to a PBAT content of 20%.25 Similarly, in the present study there was no significant effect on the crystallinity of PHA except at high contents of PBAT. The crystallization temperature of P(3HB) increased with increasing fraction of PBAT up to 60 wt% PBAT, but then decreased for 80 wt% PBAT (Fig. 5a). In contrast, the values of Tm were not affected by the blending. P(3,4HB) displayed two melting peaks which typically correspond to a melting–recrystallization–remelting process, (ESI, Fig. S7†).36 The two crystalline melting points of P(3,4HB) decreased with increasing fraction of PBAT. In addition, the crystallization temperature of P(3,4HB) decreased with increasing weight fraction of PBAT. Hence, the addition of PBAT retarded the crystallization of P(3,4HB). The same phenomenon was observed for PBAT. TGA and DSC traces and corresponding data for P(3,4HB) blends are found in ESI, Fig. S6 and S7, and Table S1.†
:PBAT blends
| P(3HB):PBAT (wt:wt) |
T PHAd/TPBATd (°C) | T PHAm/TPBATm (°C) | T PHAc/TPBATc (°C) | T PHAg/TPBATg (°C) |
|---|---|---|---|---|
| 100:0 |
291/— | 177/— | 122/— | 2/— |
| 80:20 |
292/387 | 176/— | 126/78 | 9/−34 |
| 60:40 |
290/388 | 177/— | 130/77 | 6/−31 |
| 40:60 |
289/390 | 177/123 | 131/79 | 4/−31 |
| 20:80 |
291/391 | 176/124 | 126/80 | 5/−29 |
| 0:100 |
—/391 | —/123 | —/81 | —/−29 |
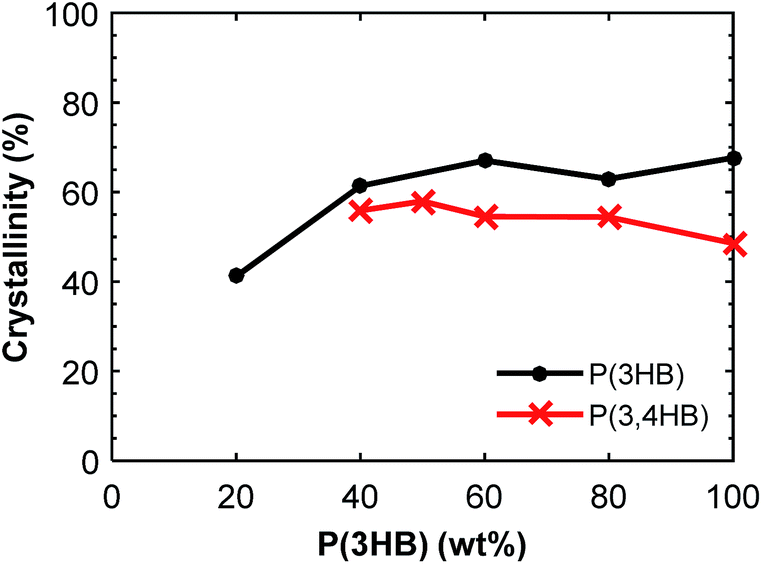 | ||
| Fig. 4 Crystallinity of the PHA fraction of the blends calculated from the second DSC heating scans. | ||
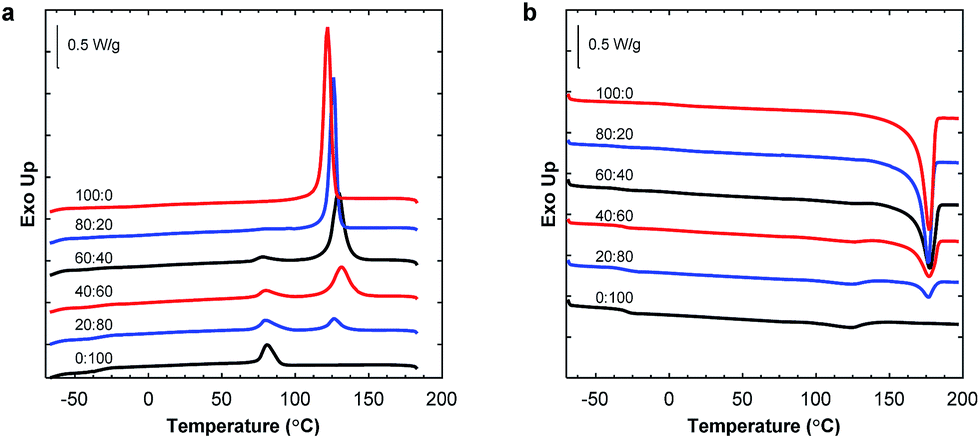 | ||
| Fig. 5 DSC traces of the P(3HB):PBAT blends: cooling (a) and second heating scans (b). | ||
:PBAT blends after measuring for 40 min, as seen in Fig. 6a. The decrease in modulus during 40 min was less for samples containing 0–60 wt% P(3HB) than for those with 80 and 100 wt% P(3HB). In addition, the samples with lower P(3HB) content reached a plateau after 15 min. Thus, there was a distinct difference between the blends containing 60 and 80 wt% P(3HB) indicating a phase inversion in this composition range. Above 60 wt% P(3HB) the differences between the samples were lesser because the more thermally stable PBAT formed the matrix phase. The large drop in modulus for the samples containing 80 and 100 wt% P(3HB) indicated thermal decomposition of P(3HB). However, it is not likely that the decomposition of P(3HB) declined for the other blends but rather that the continuous PBAT phase stabilized the modulus. Samples containing P(3,4HB) and PBAT showed similar results with a phase inversion between 60 and 80 wt% P(3,4HB) (Fig. 6b). However, the drop in the storage modulus was far less than for the P(3HB) blends. This was most probably due to the lower measurement temperature of the P(3,4HB) samples, leading to less degradation (Fig. 1b).
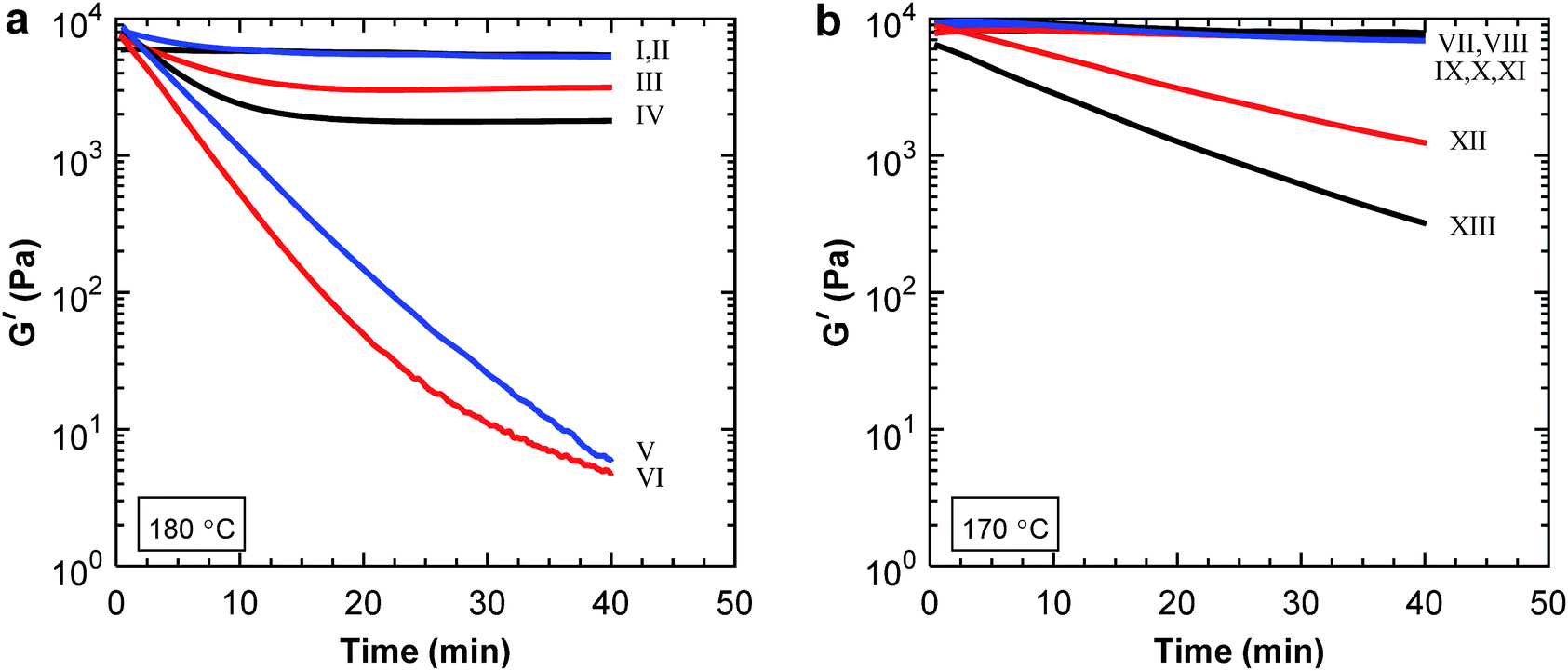 | ||
| Fig. 6 Shear storage modulus measured by rheology at the processing temperatures of (a) P(3HB):PBAT blends [0:100 (I), 20:80 (II), 40:60 (III), 60:40 (IV), 80:20 (V) and 100:0 (VI)], and (b) P(3,4HB):PBAT blends [0:100 (VII), 20:80 (VIII), 40:60 (IX), 50:50 (X), 60:40 (XI), 80:20 (XII) and 100:0 (XIV)]. | ||
The tensile storage modulus for the P(3HB):PBAT blends determined by DMA increased with increasing weight fraction of P(3HB), as seen in Fig. 7a. From the tanδ curves presented in Fig. 7b it was apparent that the damping increased with increasing fraction of PBAT, which has a higher amorphous content. P(3HB) is a stiffer material than PBAT at room temperature, and the results from the DMA measurements confirmed this difference in material properties. The immiscibility was indicated by two separate peaks appearing in the loss modulus, corresponding to the P(3HB) and PBAT glass transitions, respectively. As seen in the inset of Fig. 7b, the Tg of PBAT decreased somewhat with increasing fraction of P(3HB) which was consistent with the DSC results. However, the Tg of P(3HB) decreased with increasing PBAT fraction, which was not observed in the DSC data. Considering the blends of P(3,4HB) and PBAT, the same trends of G′ and tanδ was observed (ESI, Fig. S9†). However, the Tg values of P(3,4HB) slightly increased with increasing fraction of PBAT up to 40 wt% PBAT and thereafter a decrease in Tg was observed.
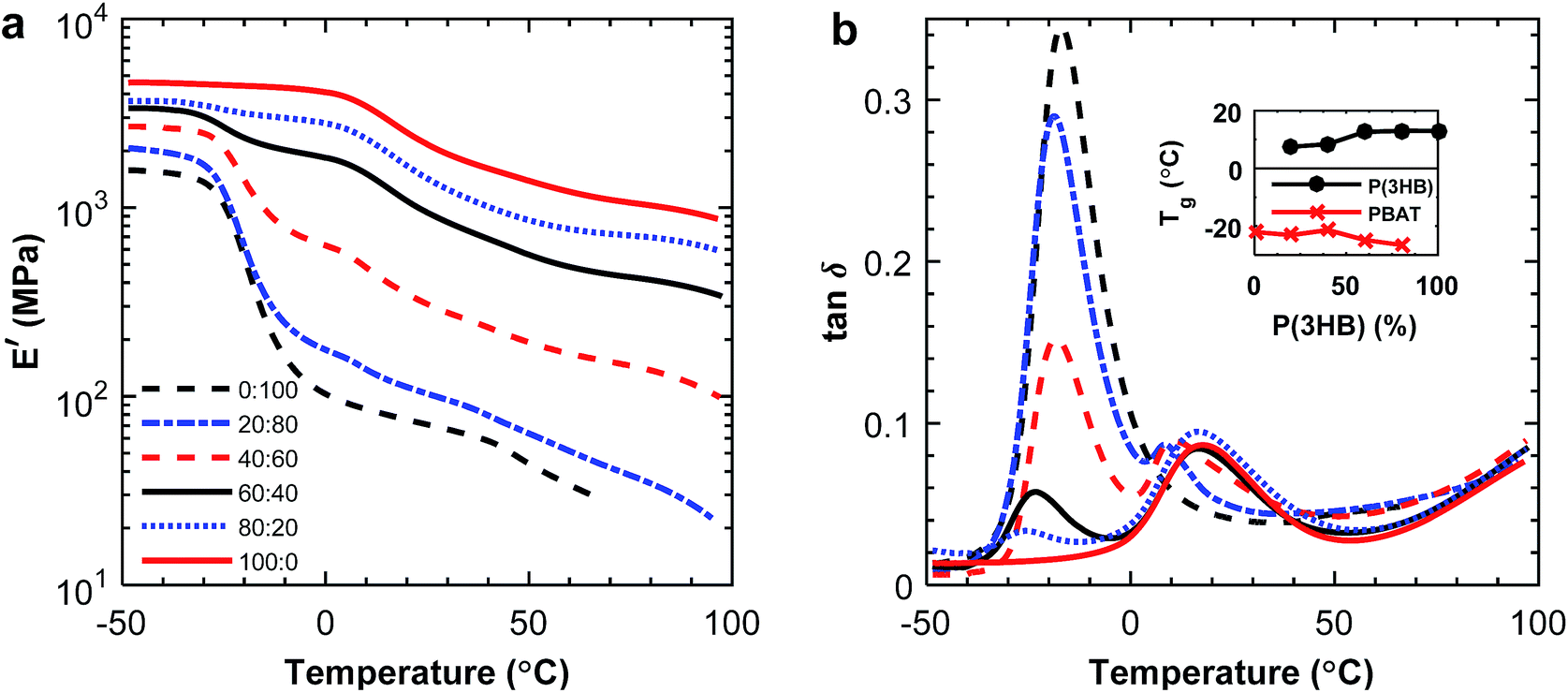 | ||
| Fig. 7 Tensile storage modulus (a) and tanδ (b) of the P(3HB):PBAT blends. The inset presents the Tg of P(3HB) and PBAT in the blends determined from the position of the loss modulus peak. | ||
Reactive extrusion
In order to increase the compatibility between the immiscible polymers in blends, a compatibilizer may be added in situ to perform a reactive extrusion. Dicumyl peroxide (DCP) has previously been used as a compatibilizer in P(3HB)/PBS and P(3HB)/PLA blends. Consequently, the phase domain sizes decreased while the elongation at break, impact toughness and tensile strength was increased.19,32 Wei and McDonald studied reactive extrusion of P(3HB) and PLA, respectively, with DCP as a crosslinking agent, and concluded that the crosslinked materials showed an improved melt strength.37In the present study two especially interesting blend compositions were chosen for the reactive extrusion experiments. The 20:80 blend composition was selected because PBAT was the continuous phase and P(3HB) may then potentially act as a barrier material in a soft and strong matrix as described above. In addition, the 60:40 blend was chosen since PBAT still appeared to be the continuous phase with a stable modulus according to the rheology measurements. Samples containing 1 wt% DCP were found to be difficult to process due to the high viscosity caused by a rapid reaction. Due to this issue, DCP was added to the extruder together with the very last portion of polymer. DCP has a half-life time of approx. 0.5 min at 180 °C and it was assumed that the DCP was completely decomposed during the processing.38
In addition to the blends, P(3HB) and PBAT were separately extruded with DCP. In the case of P(3HB) the reaction was rapid, indicated by a sharp increase of the torque. In contrast, there was no immediate effect observed for PBAT. A solubility study of the extruded materials revealed the formation of an extensive crosslinked gel fraction for P(3HB). No effect was observed for PBAT which remained soluble. Thus, it was likely that the reaction in the blends mainly affected the P(3HB) phase because of its tertiary hydrogens, which are readily abstracted by the primary radical formed by DCP.
:80 blends. In the non-compatibilized blend there were discrete phase domains of P(3HB) clearly visible in a continuous PBAT matrix. In contrast, individual phase domains were not visible in the compatibilized blend, which displayed a rather smooth surface. These observations indicated that the addition of DCP significantly improved the compatibility of the blend components.
 | ||
| Fig. 8 SEM images of cryo-fractured surfaces of P(3HB):PBAT 20:80 blends (a) without DCP and (b) with 0.5 wt% DCP added. | ||
In order to further investigate the adhesion between the phases, the blend containing 60 wt% of P(3HB) was fractured at room temperature. The sample with a larger content of PBAT was more difficult to fracture in this way due to the high elongation at break of PBAT. There was a difference in the phase morphology at the surface between the samples without DCP and with 0.5 wt% DCP added (Fig. 9). As seen in the former case, the PBAT domains were clearly deformed compared to the latter, where the effect was less visible. Again, this indicated an increased compatibility between the polymer phases.
 | ||
| Fig. 9 SEM images of room temperature fractured surfaces of P(3HB):PBAT 60:40 blends (a) without DCP and (b) with 0.5 wt% DCP added. | ||
:80 blend composition, from 175 to 168 °C when 0.75 wt% DCP was added. The same trend was observed for the blend with 60:40 blend ratio. The results confirmed a complete consumption and decomposition of DCP during the extrusion.
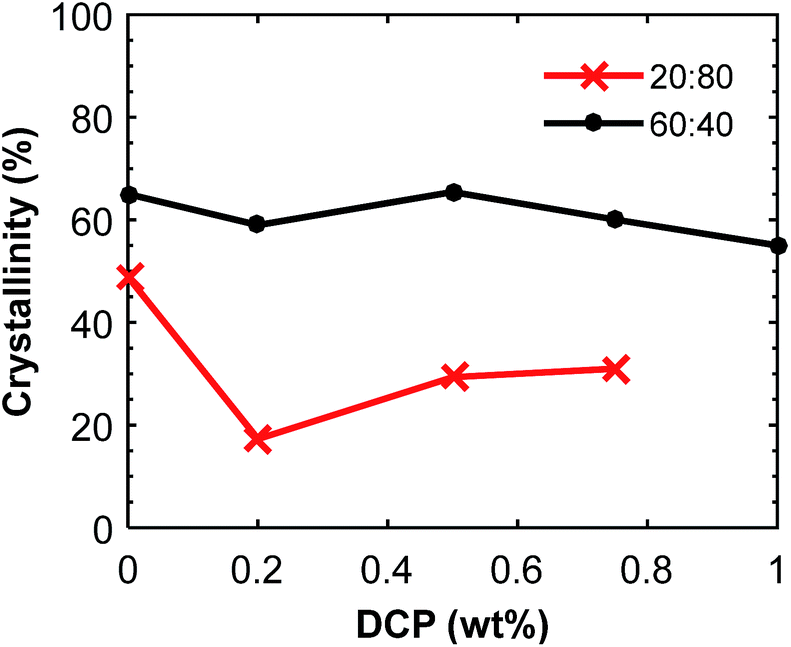 | ||
| Fig. 10 Crystallinity of the P(3HB) component in P(3HB):PBAT blends as a function of the DCP content. The data was calculated from the second DSC heating scans (ESI, Fig. S11 and S12†). | ||
:40 blend a similar increase of the modulus and decrease of the phase shift with increasing content of DCP was observed (ESI, Fig. S13†). It is important to consider the complexity of this system. The changes in properties depend on the respective polymer's molecular weight (i.e. affected by the thermal decomposition of PHA), morphology and the crosslinking by peroxide.
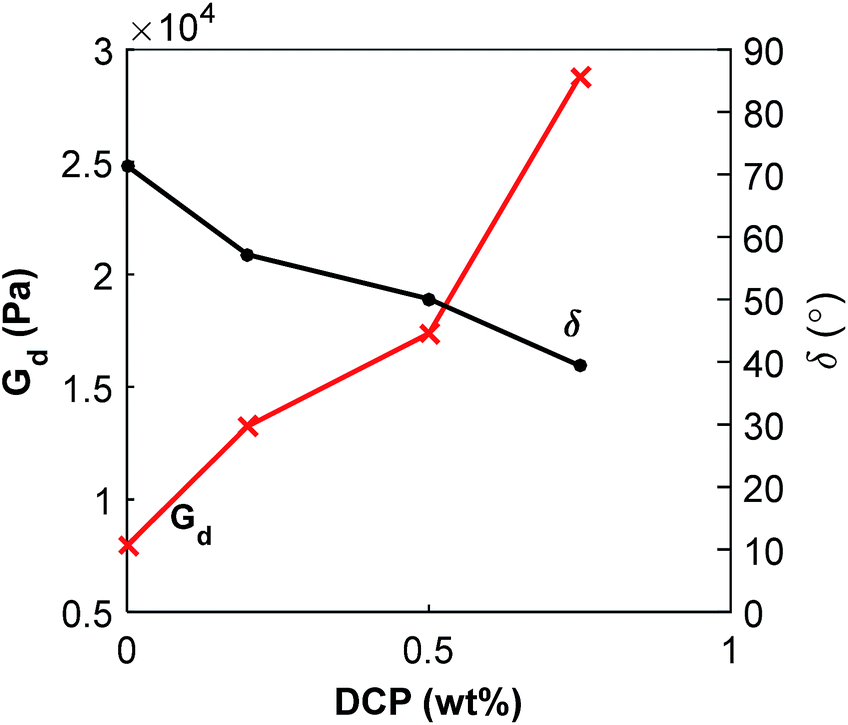 | ||
| Fig. 11 Dynamic shear modulus and phase shift for P(3HB):PBAT 20:80 blends with addition of DCP measured by rheology at 1 Hz and 180 °C. | ||
The effect of compatibilization was also visible in the tensile storage modulus measured by DMA which increased by 48% at 20 °C for the sample with 0.5 wt% DCP added (Fig. 12a). In contrast, the tensile storage modulus for the 60:40 blends at 20 °C decreased after additions of DCP, in comparison with the blend without DCP (ESI, Fig. S14a†). Considering the damping, there was a small decrease in the tanδ peak with the addition of DCP, which indicated a stiffer material (Fig. 12b). Also, there were two maxima visible in both the tanδ and the loss modulus, relating to the glass transitions of respective polymer. In the 60:40 blend the tanδ peak of P(3HB) increased with the addition of DCP (ESI, Fig. S14b†).
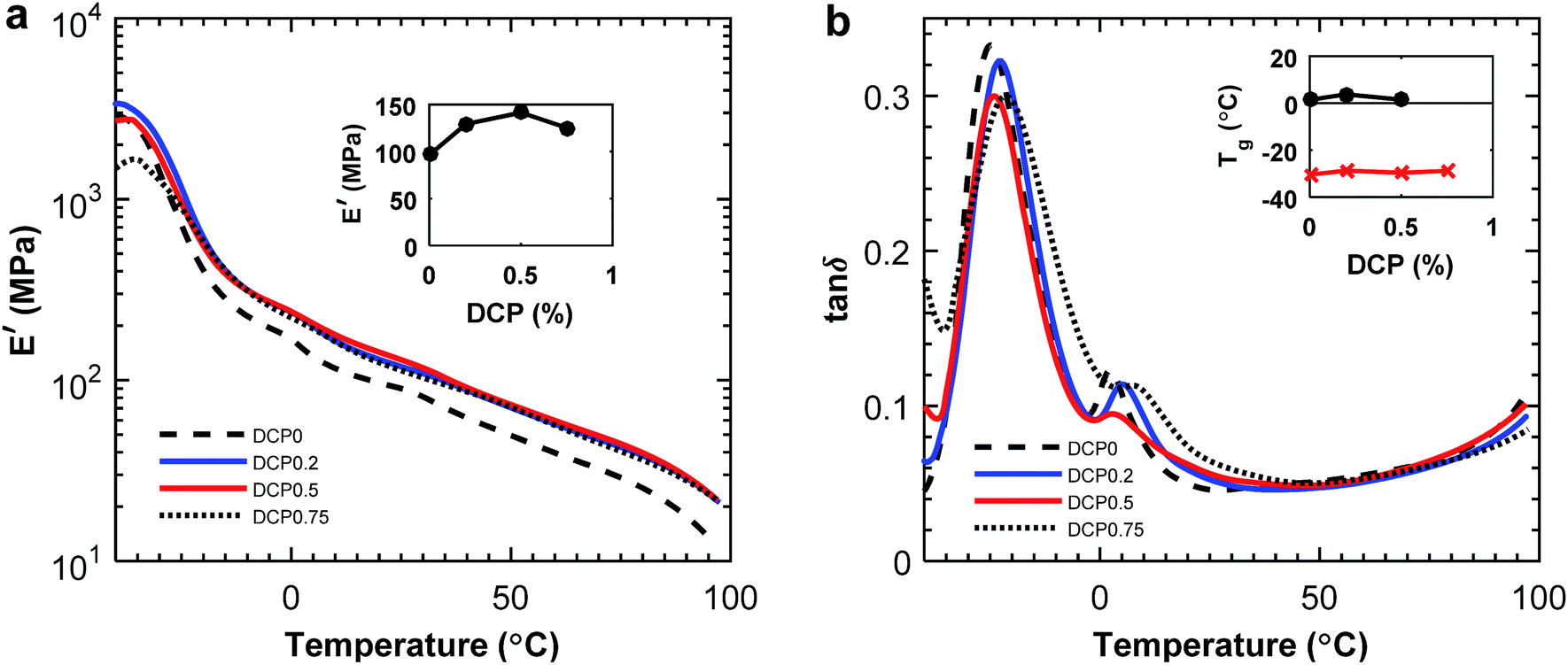 | ||
Fig. 12 Tensile storage modulus (a) and tanδ (b) for P(3HB):PBAT 20:80 blends with addition of DCP. The inset in part (a) shows the change in E′ with the content of DCP, and the inset in part (b) shows the Tg of P(3HB) ( ) and PBAT ( ) and PBAT ( ) determined from the loss modulus peak value. ) determined from the loss modulus peak value. | ||
Conclusions
When melt processing PHBs from commercial sources it is crucial to assess and, if necessary, improve the thermal stability. In the present case, washing with aq. HCl solution was identified as the most efficient and environmentally friendly pre-treatment method. The thermal decomposition temperature of PHA increased by 50 °C and the loss of molecular weight during processing was considerably less severe after the acid treatment. Blending PHA with a suitable biodegradable polymer is an efficient strategy to improve the processability. The shear storage modulus of the blends increased with increasing content of PBAT and the stability in the melt state was higher for the samples where PBAT formed the continuous phase. Moreover, the tensile storage modulus decreased and the brittleness was reduced when PBAT was added. Addition of a free radical initiator such as DCP provided compatibilization between the polymers. This resulted in an increase of the dynamic shear modulus, an apparent increase in interfacial adhesion, an increase in tensile storage modulus, and can be effectively used to tune the properties of the materials.Acknowledgements
We gratefully acknowledge The Swedish Research Council and Baxter for financial support, and Reine Wallenberg for advice on electron microscopy and Frans Maurer for valuable discussions.Notes and references
- E. Bugnicourt, P. Cinelli, A. Lazzeri and V. Alvarez, eXPRESS Polym. Lett., 2014, 8, 791–808 CrossRef.
- A. Tsui, Z. C. Wright and C. W. Frank, Annu. Rev. Chem. Biomol. Eng., 2013, 4, 143–170 CrossRef CAS.
- S. Y. Lee, Biotechnol. Bioeng., 1996, 49, 1–14 CrossRef CAS PubMed.
- N. Grassie, E. J. Murray and P. A. Holmes, Polym. Degrad. Stab., 1984, 6, 95–103 CrossRef CAS.
- W. M. Pachekoski, C. Dalmolin and J. A. M. Agnelli, Materials Research–Ibero-american Journal of Materials, 2013, 16, 327–332 CAS.
- L. Montano-Herrera, S. Pratt, M. V. Arcos-Hernandez, P. J. Halley, P. A. Lant, A. Werker and B. Lancock, New Biotechnol., 2014, 31, 357–363 CrossRef CAS.
- R. Renstad, S. Karlsson and A. C. Albertsson, Polym. Degrad. Stab., 1997, 57, 331–338 CrossRef CAS.
- A. El-Hadi, R. Schnabel, E. Straube, G. Muller and M. Reimschneider, Macromol. Mater. Eng., 2002, 287, 363–372 CrossRef CAS.
- S. Gogolewski, M. Jovanovic, S. M. Perren, J. G. Dillon and M. K. Hughes, Polym. Degrad. Stab., 1993, 40, 313–322 CrossRef CAS.
- C. Thellen, M. Coyne, D. Froio, M. Auerbach, C. Wirsen and J. A. Ratto, J. Polym. Environ., 2008, 16, 1–11 CrossRef CAS.
- M. Kunioka, A. Tamaki and Y. Doi, Macromolecules, 1989, 22, 694–697 CrossRef CAS.
- K. Ishida, Y. Wang and Y. Inoue, Biomacromolecules, 2001, 2, 1285–1293 CrossRef CAS PubMed.
- X. Wen, X. P. Lu, Q. Peng, F. Y. Zhu and N. Zheng, J. Therm. Anal. Calorim., 2012, 109, 959–966 CrossRef CAS.
- I. Armentano, E. Fortunati, N. Burgos, F. Dominici, F. Luzi, S. Fiori, A. Jiménez, K. Yoon, J. Ahn, S. Kang and J. M. Kenny, eXPRESS Polym. Lett., 2015, 9, 583–596 CrossRef CAS.
- M. A. Abdelwahab, A. Flynn, B. S. Chiou, S. Imam, W. Orts and E. Chiellini, Polym. Degrad. Stab., 2012, 97, 1822–1828 CrossRef CAS.
- M. P. Arrieta, E. Fortunati, F. Dominici, E. Rayon, J. Lopez and J. M. Kenny, Polym. Degrad. Stab., 2014, 107, 139–149 CrossRef CAS.
- M. Zhang and N. L. Thomas, Adv. Polym. Technol., 2011, 30, 67–79 CrossRef CAS.
- D. D. Ju, L. J. Han, J. J. Bian, Z. Q. Guo, F. Li, S. Chen and L. Dong, RSC Adv., 2015, 5, 5474–5483 RSC.
- P. M. Ma, D. G. Hristova-Bogaerds, P. J. Lemstra, Y. Zhang and S. F. Wang, Macromol. Mater. Eng., 2012, 297, 402–410 CrossRef CAS.
- F. Gassner and A. J. Owen, Polymer, 1994, 35, 2233–2236 CrossRef CAS.
- P. Mousavioun, P. J. Halley and W. O. S. Doherty, Ind. Crops Prod., 2013, 50, 270–275 CrossRef CAS.
- A. Javadi, A. J. Kramschuster, S. Pilla, J. Lee, S. Q. Gong and L. S. Turng, Polym. Eng. Sci., 2010, 50, 1440–1448 CAS.
- P. Dacko, M. Kowalczuk, H. Janeczek and M. Sobota, Macromol. Symp., 2006, 239, 209–216 CrossRef CAS.
- Y. Parulekar and A. K. Mohanty, Macromol. Mater. Eng., 2007, 292, 1218–1228 CrossRef CAS.
- L. Jiang, M. P. Wolcott and J. W. Zhang, Biomacromolecules, 2006, 7, 199–207 CrossRef CAS PubMed.
- D. F. Wei, H. Wang, H. N. Xiao, A. N. Zheng and Y. Yang, Carbohydr. Polym., 2015, 123, 275–282 CrossRef CAS PubMed.
- J. A. Stagner, V. D. Alves and R. Narayan, J. Appl. Polym. Sci., 2012, 126, E135–E142 CrossRef CAS.
- R. Muthuraj, M. Misra and A. K. Mohanty, J. Polym. Environ., 2014, 22, 336–349 CrossRef CAS.
- J. Yu and L. X. L. Chen, Biotechnol. Prog., 2006, 22, 547–553 CrossRef CAS PubMed.
- (a) C. R. Arza, P. Jannasch and F. H. J. Maurer, Eur. Polym. J., 2014, 59, 262–269 CrossRef CAS; (b) A. Werker, P. Johansson, P. Magnusson, F. H. J. Maurer and P. Jannasch, Patent WO 2012022998, 2012.
- M. Lopez-Abelairas, M. Garcia-Torreiro, T. Lu-Chau, J. M. Lema and A. Steinbuchel, Biochem. Eng. J., 2015, 93, 250–259 CrossRef CAS.
- W. F. Dong, P. M. Ma, S. F. Wang, M. Q. Chen, X. X. Cai and Y. Zhang, Polym. Degrad. Stab., 2013, 98, 1549–1555 CrossRef CAS.
- P. J. Barham, A. Keller, E. L. Otun and P. A. Holmes, J. Mater. Sci., 1984, 19, 2781–2794 CrossRef CAS.
- C. R. Arza, P. Jannasch, P. Johansson, P. Magnusson, A. Werker and F. H. J. Maurer, J. Appl. Polym. Sci., 2015, 132, 41836, DOI:10.1002/app.41836.
- K. Csomorova, J. Rychly, D. Bakos and I. Janigova, Polym. Degrad. Stab., 1994, 43, 441–446 CrossRef CAS.
- P. J. Pan, Z. C. Liang, N. Nakamura, T. Miyagawa and Y. Inoue, Macromol. Biosci., 2009, 9, 585–595 CrossRef CAS PubMed.
- L. Q. Wei and A. G. McDonald, J. Appl. Polym. Sci., 2015, 132, 41724, DOI:10.1002/app.41724.
- T. Chatterjee, S. Wiessner, K. Naskar and G. Heinrich, eXPRESS Polym. Lett., 2014, 8, 220–231 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data and graphs. See DOI: 10.1039/c6ra06282b |
| This journal is © The Royal Society of Chemistry 2016 |