
Fluorescent, online monitoring of PLGA degradation for regenerative medicine applications†
Abstract
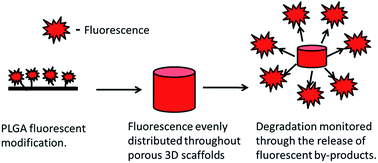
Degradable polymers such as poly(lactic-co-glycolic acid) (PLGA) are frequently chosen for tissue engineering, due to their ease of production, controllable degradation rates and Food and Drug Administration (FDA) approval. Within tissue engineering it is essential that the degradation profile of such biomaterials is understood and measured both in vitro and in vivo. The majority of techniques currently undertaken to study degradation are however destructive, leading to an over reliance on end point analysis. This study therefore defines a method of fluorescently tagging PLGA, via the addition of reactive amine groups and subsequent isothiocyanate reactions, with the purpose of monitoring degradation profiles through non-destructive techniques. The amine grafting and fluorescent labelling of the PLGA was confirmed using both X-ray photospectrometry and high performance liquid chromatography. The modification of the PLGA also had no significant effect on molecular weight or the hydrophilicity of the polymer. Both the release of fluorescent by-products and the changes in fluorescence retention within the modified PLGA were observed to be highly correlated to the changes in physical weight. This paper therefore demonstrates a novel method for the online and non-destructive monitoring of polymer degradation through the incorporation of a fluorescent marker, which can decrease the reliance on end point analysis and reduce the number of samples required both in vitro and in vivo.
 Please wait while we load your content...
Please wait while we load your content...

