
Selective detection of carbon monoxide (CO) gas by reduced graphene oxide (rGO) at room temperature†
Abstract
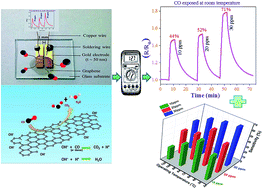
Graphene materials have been widely explored for fabrication of gas sensors because of their atom-thick two-dimensional conjugated structures, high conductivity and large specific surface area. Thin graphene layers with attached functional groups desirable for gas sensing applications were synthesized by a wet chemical route (modified Hummers' method). Sensing performances of reduced graphene oxide (rGO) against carbon monoxide (CO) were studied in terms of percent sensitivity (sensor response), response and recovery times and I/V characteristics at room temperature as well as at elevated temperatures. Sensitivity data indicate the highest activity (∼71% sensitivity against 30 ppm CO) occurs at room temperature (RT), indicating that the sensor is best operated at RT. Sensor response is quick (within 30 s) even to a trace amount (0.001% or, 10 ppm) of CO gas. Selectivity of the sensor was demonstrated by using different n-type reducing gases (like carbon monoxide, ammonia, methane and hydrogen), at different concentrations, showing negligible sensitivity towards gases other than CO. The synthesized material proves to be good as a selective room temperature sensor for harmful and poisonous carbon-monoxide gas.
 Please wait while we load your content...
Please wait while we load your content...

