
Polyacrylamide strengthened mixed-charge hydrogels and their applications in resistance to protein adsorption and algae attachment
Abstract
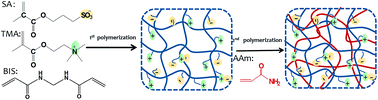
Mixed-charge polymer hydrogels were successfully prepared by copolymerization of different ratios of [2-(meth-acryloyloxy) ethyl] trimethylammonium (TMA) and 3-sulfopropyl methacrylate (SA). Then, a second polyacrylamide (PAAM) network was incorporated into the pre-prepared hydrogel to form a double network (DN) hydrogel. The compositions of these DN hydrogels were characterized by Fourier transform infrared spectroscopy (FTIR) and X-ray photoelectron spectroscopy (XPS). Rheological and compressive measurements confirmed that the mechanical performances of the DN hydrogels were significantly improved by incorporation of a second PAAM network, compared with the according single network (SN) hydrogels. The amount of protein absorbed on the DN hydrogel surface was related to the ratio of TMA/SA and the ionic strength. The DN hydrogel with equal amount of TMA and SA exhibited better protein resistance. In addition, Phaeodactylum tricornutum and Chlorella were chosen for the anti-algae assay. The results displayed that the negatively charged hydrogels showed better anti-algae fouling performance than the positively charged and the neutral DN hydrogels. These DN hydrogels have promising applications in marine antifouling coating and interfaces of biomaterials.
 Please wait while we load your content...
Please wait while we load your content...

