
Alkylated histidine based short cationic antifungal peptides: synthesis, biological evaluation and mechanistic investigations†
Abstract
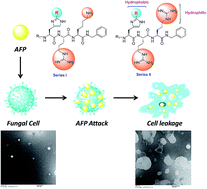
Current clinically used antifungal agents suffer from several drawbacks that have urgently necessitated the development of new antifungal agents with unusual mechanisms of action. In this context, antifungal peptides (AFPs) open up new perspectives in drug design by providing an entire range of highly selective and nontoxic pharmaceuticals. Here, we report the development of novel short AFPs with the synthesis of two series of tripeptide based compounds named as His(2-alkyl)-Arg-Lys (series I) and His(2-alkyl)-Arg-Arg (series II). The series II peptides were found to be selectively active against Cryptococcus neoformans whereas some peptides displayed encouraging activities against other fungal strains such as Candida albicans, Candida kyfer, Aspergillus niger and Neurospora crassa. The cytotoxic experiments were performed on active compounds using Hek-293 and HeLa cells which exhibited negligible cytotoxic effect up to the highest test concentration. Further, the most potent peptide was subjected to mechanistic studies using TEM analysis. Two sets of SUVs mimicking microbial membrane and mammalian membrane were treated with the most potent peptide. The results of this study were found to be perfectly in corroboration with the antifungal activity in relation to the differences between microbial and mammalian cell membrane composition, thereby, indicating that the reported peptides may also be less susceptible to the common mechanisms of drug resistance.
 Please wait while we load your content...
Please wait while we load your content...

