
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel trinitroethanol derivatives: high energetic 2-(2,2,2-trinitroethoxy)-1,3,5-triazines†
Alexander A.
Gidaspov
a,
Vladimir A.
Zalomlenkov
a,
Vladimir V.
Bakharev
a,
Victor E.
Parfenov
a,
Evgeniy V.
Yurtaev
a,
Marina I.
Struchkova
b,
Nadezhda V.
Palysaeva
b,
Kyrill Yu
Suponitsky
c,
David B.
Lempert
d and
Aleksei B.
Sheremetev
*b
aSamara State Technical University, Samara, 443100, Russian Federation
bN. D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, 47 Leninsky Prosp, Moscow 119991, Russian Federation
cA. N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Science, Moscow 119991, Russian Federation
dInstitute of Problems of Chemical Physics, Russian Academy of Sciences, Chernogolovka, Moscow Region, 142432 Russian Federation
First published on 1st April 2016
Abstract
The multicomponent reaction of 2,4,6-trichloro-1,3,5-triazine with potassium trinitromethane and trinitroethanol was exploited for the first synthesis of the hetaryl trinitroethyl ether, 2,4-bis(2,2,2-trinitroethoxy)-6-trinitrometyl-1,3,5-triazine 13. The use of compound 13 as a scaffold for the synthesis of substituted trinitroethoxytriazine by sequential nucleophilic substitution processes is described. A number of trinitroethoxytriazines bearing a range of functional groups, including 2,4,6-tris(2,2,2-trinitroethoxy)-1,3,5-triazine 16, have been prepared. There has been no previous incorporation of the trinitroethoxy moiety to a heteroaromatic ring. All trinitroethoxytriazines were fully characterized using IR and multinuclear NMR spectroscopy, elemental analysis, and differential scanning calorimetry (DSC), and, in some cases, 16, 20 and 21, with single crystal X-ray structuring. When compared to the aliphatic trinitroethoxy compounds, the trinitroethoxytriazines show better energetic performance as calculated. The impact sensitivities and ignition points of the novel oxygen and nitrogen-rich triazines were measured. The ability of the applied trinitroethoxytriazines in solid composite propellants as well as in gas generant compositions for airbag inflators was evaluated. The straightforward preparation of these ethers highlights them as valuable new environmentally friendly and high-performing nitrogen and oxygen-rich materials.
Introduction
Heterocyclic compounds have found many applications in all parts of the chemical industry. In particular, heterocyclic chemistry is of central importance to energetic materials creation and the majority of newly prepared high energy compounds contain at least one heterocyclic subunit within their structures.1 Nitrogen-heterocycles generally possess performance-like properties, such as high density, high enthalpy of formation, and favorable oxygen balance. There is a continued current demand for the development of structurally new heterocyclic architectures2 to obtain novel, valuable energetic and physical properties.A large number of 1,3,5-triazines bearing a wide range of explosophoric groups, namely –NHNO2,3 –N3,4 –NHCH2C(NO2)3,5 –C(NO2)3,6 –Het(NO2),7 and others,8 have found applications in a significant number of energetic compounds and some examples, including a nitro alcohol derivative9 relevant to the chemistry described in this paper, are shown in Fig. 1. Progress in the synthesis and chemistry of energetic 1,3,5-triazines has been the subject of recent reviews.10
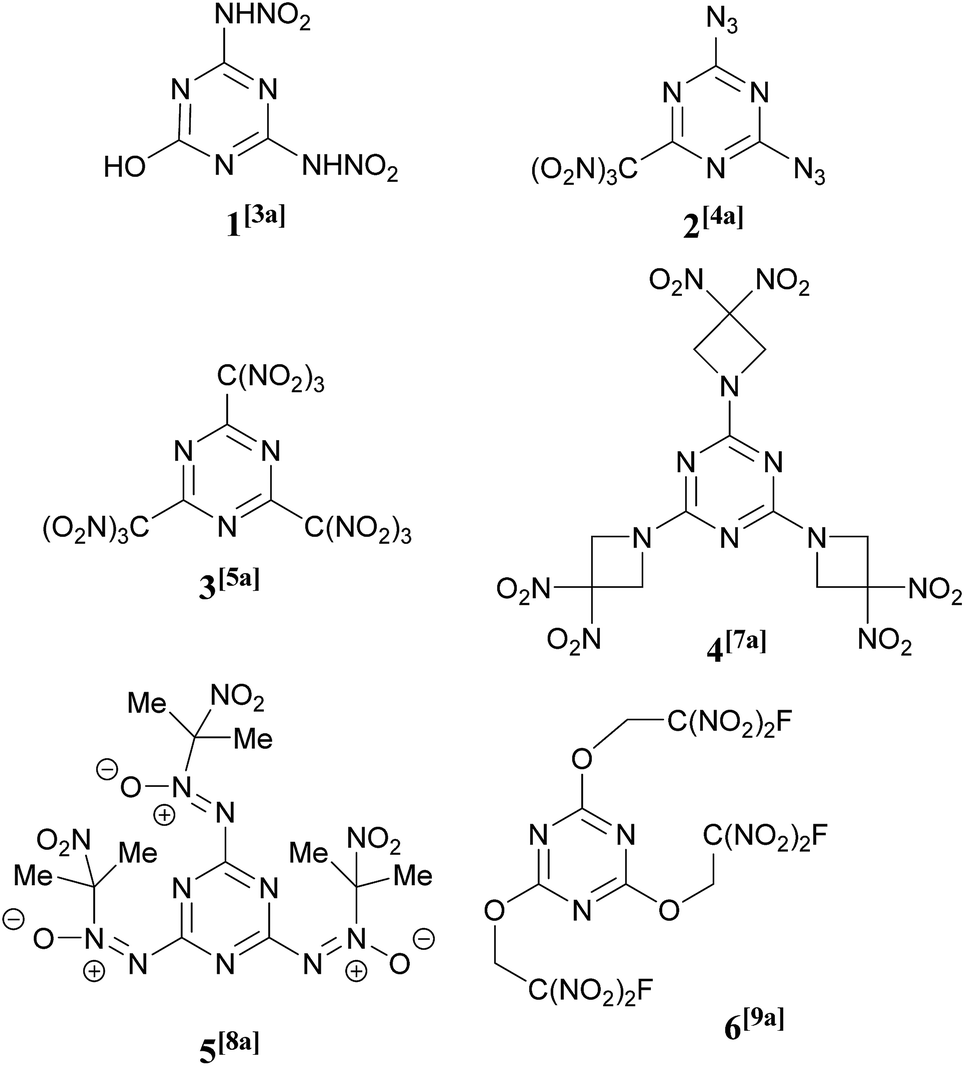 | ||
| Fig. 1 Energetic 1,3,5-triazine derivatives. | ||
Polynitro compounds are of significant interest as oxygen-rich energetic materials for propulsion and explosive applications.1 2,2,2-Trinitroethanol has long been recognized as a useful building block for such materials. Progress in the synthesis and chemistry of the electro negatively substituted alcohol and its analogues has been the subject matter of monographs11 and reviews.12 However, the reactivity of trinitroethanol is much lower than that of usual alcohols and, in some cases, is very specific. Thus, at pH values greater than 6, the retro-Henry reaction has been observed and a mixture of the trinitromethanide anion and formaldehyde was formed.11,12 As a result, attempts to apply the trinitroethanol as the oxygen centered nucleophile for halide displacement in SNAr reaction at traditional conditions failed to yield any of the expected (het)aryl trinitroethyl ether since the alcohol decomposed in the presence of a base.
The only reported examples of trinitroethanol ethers of any cycle are 2-(2,2,2-trinitroethoxy)-4-(polynitromethyl)-tetrahydrofurans13 formed from 4-(polynitromethyl)-tetrahydro-2-furanol and the nitroalcohol in the presence of an acid catalyst with azeotropic removal of water, and 2-exo-(2,2,2-trinitroethoxy)norbornanes14 formed from norbornene and trinitroethanol in the presence of XeF2. No previous aryl or heteroaryl trinitroethyl ethers are reported.
In this paper, we report our studies aimed at developing unprecedented method for manipulation of trinitroethanol as the nucleophile. Herein, synthetic routes to the unique scaffold comprised of 1,3,5-triazine and 2,2,2-trinitroethoxy moieties are presented. Connection these two excellent building blocks produces uniquely functionalized molecular architecture that could find utility as a scaffold for the construction of a range of energetic compounds.
Results and discussion
Synthesis
In a developing research program, we reported a multicomponent nucleophilic displacement reaction using a combination of 2,4,6-trichloro-1,3,5-triazine, trinitromethane salts, and a nucleophile (optimized conditions: a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:4, respectively, the concentration of the trinitromethane salt solution was 2.7 M in acetone, at room temperature), which provides rapid access to trinitromethyl containing triazines.15 The short and efficient method for the preparation of energetic materials is illustrated in Scheme 1. During the course of previous studies,15 we also observed the formation of hydroxytriazines, which are side products that result from the hydrolysis of intermediated nitronic esters.
:4:4, respectively, the concentration of the trinitromethane salt solution was 2.7 M in acetone, at room temperature), which provides rapid access to trinitromethyl containing triazines.15 The short and efficient method for the preparation of energetic materials is illustrated in Scheme 1. During the course of previous studies,15 we also observed the formation of hydroxytriazines, which are side products that result from the hydrolysis of intermediated nitronic esters.
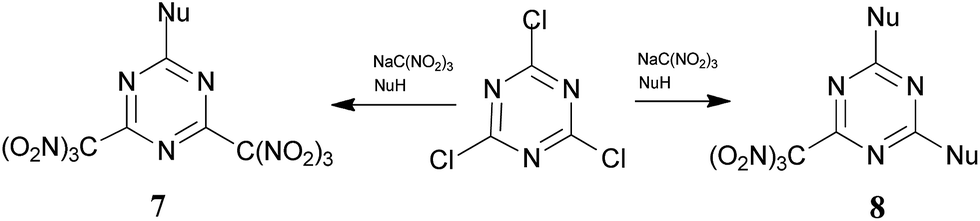 | ||
| Scheme 1 | ||
Although the reactions were successful using such alcohols as 2-fluoro- and 2-chloro-2,2-dinitroethanols as nucleophile synthons (NuH), no characterizable product(s) could be obtained when trinitroethanol was exposed to the same reaction conditions: exothermic reaction in the presence of the alcohol was observed, and no detectable product was formed.
The combination of trinitroethanol and trinitromethane salt presents the following challenges: (i) trinitroethanol is a very weak nucleophile, and (ii) nitronic ester intermediates are unstable, and quickly decompose under the reaction conditions. As a result, the intermediates decomposed before trinitroethanol had time to react. We considered that these problems might be mitigated by use of low concentrations of the trinitromethane salt solution.
Our attempts to couple 2,4,6-trichloro-1,3,5-triazine with trinitroethanol and sodium trinitromethanide (solubility of the salt is ca. 55 g per 100 mL of acetone) using the same ratio (1:4:4) at low concentrations of all reactants in acetone (0.2 M, 0.8 M and 0.8 M, respectively) met with no success. Then we prepared more concentrated solution of 2,4,6-trichloro-1,3,5-triazine with trinitroethanol (0.8 M and 3.2 M, respectively) and sodium trinitromethanide was added in several portions. Different numbers of portions were tested to find optimal conditions. In the best experiment, 4 equiv. of sodium trinitromethanide was added in eight portions over a period of 2 days at room temperature, giving the first known hetaryltrinitroethyl ether, compound 13 (Scheme 2), in moderate yield (32%). Neither longer reaction time nor lower temperature increased the yield of the multicomponent reaction product. Moreover, it should be noted that it was necessary to use freshly prepared unstable (and therefore unusable) sodium trinitromethanide for each portion.
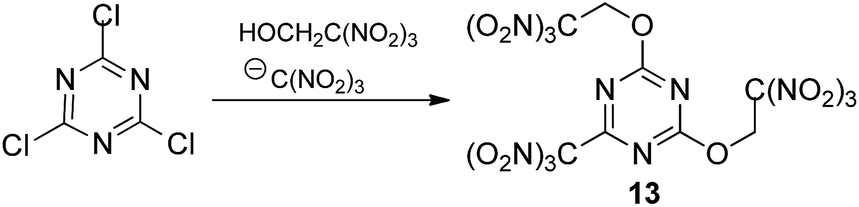 | ||
| Scheme 2 | ||
More stable and less soluble in acetone potassium trinitromethanide, KC(NO2)3, proved to be superior to sodium salt as a source of the trinitromethane anion. The ether 13 was formed in similar yield (30%), when a slurry of 2,4,6-trichloro-1,3,5-triazine with trinitroethanol (4 equiv., 3.2 M) and KC(NO2)3 (4 equiv., addition in one portion) in acetone were stirred at room temperature during two days. Solubility of KC(NO2)3 is ca. 7 g per 100 mL of acetone at 20 °C, consequently, the concentration of the reagent does not exceed ca. 0.37 M; it slowly dissolves during the course of the reaction. Optimization experiments determined that use of additional solvent (+30%) led to more rapid consumption of 2,4,6-trichloro-1,3,5-triazine, but lower yields of product 13 (24%). Use of too little solvent (−30%) led to problems with stirring the reaction mixture.
In order to reduce the requirement for excess of trinitroethanol the reactions were also trialed in acetone with 4 equiv. of KC(NO2)3. At 20 °C good conversions were observed with only 2.5 equiv. of the alcohol to give the expected product 13 in 32% yield after 2 days. However, reducing excess of KC(NO2)3 to 3–3.5 equiv. gave the required ether 13 in 15–22% yield, together with some recovered starting 2,4,6-trichloro-1,3,5-triazine. In the absence of KC(NO2)3, 2,4,6-trichloro-1,3,5-triazine does not react with the trinitroethanol. The conditions that were eventually settled upon as optimal included using trinitroethanol in 2,5-fold excess over 2,4,6-trichloro-1,3,5-triazine, 4 equiv. of potassium trinitromethanide, a reaction temperature of 20 °C, and a time of the reaction of 2 days, which was required in order to ensure full conversion.
Attempts to displace chlorine at 2,4-dichloro-6-alkoxy-1,3,5-triazines and 2-amino-4,6-dichloro-1,3,5-triazine with trinitroethanol resulted in no reaction with the applied conditions. Trinitromethyl group can be used in place of a halogen as more effective leaving group in reactions with many nucleophiles.10a,10c Indeed, treatment of 2,4-di(trinitromethyl)-6-alkoxy-1,3,5-triazine 14 (ref. 16) with trinitroethanol in the presence of KC(NO2)3 in acetone at 20 °C gave mono-trinitroethyl ethers 15 in ca. 55% yield via displacement of one trinitromethyl group (Scheme 3). In the absence of KC(NO2)3 compound 14 does not react with trinitroethanol.
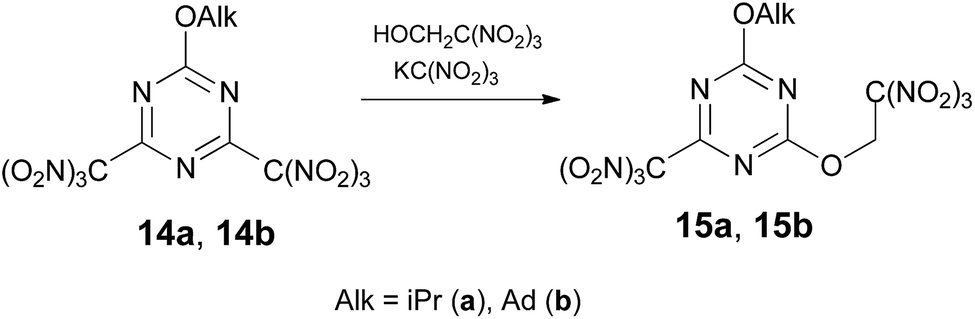 | ||
| Scheme 3 | ||
To our delight, we were able to promote by potassium trinitromethanide the displacement of trinitromethyl group in compound 13 using 5 equiv. of trinitroethanol (Scheme 4). The yield for the preparation of tris(trinitroethoxy) product 16 was 64% after 10 days. Again, KC(NO2)3 was required to the trinitroethoxylation.
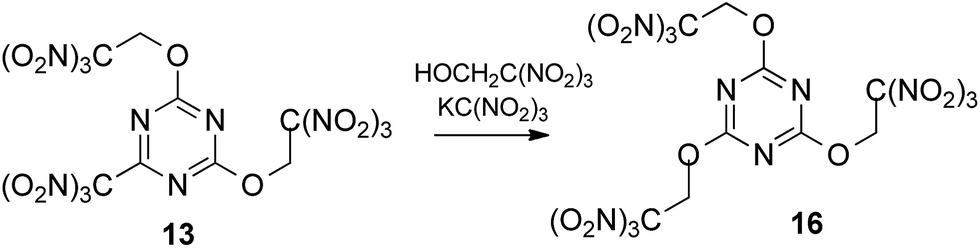 | ||
| Scheme 4 | ||
Reactions of the trinitroethoxy triazine scaffold 13 with representative nitrogen and oxygen centered nucleophiles were studied in order to provide an indication of the reactivity of the system towards nucleophilic attack and to establish the regioselectivity of such processes.
Reactions of compound 13 with sodium azide in acetonitrile and methanol in the presence of K2CO3 gave azide 17 (84%) and methoxy derivative 18 (60%) respectively, where trinitromethyl group is displaced selectively in both cases (Scheme 5). Hydroxyl nucleophile was relatively unselective in its reaction with compound 13 (Scheme 5). Both Na2CO3 and Na3PO4 gave mixtures of products 19 and 20 arising from substitution of the trinitromethyl group and the trinitroethoxyl group, which could be separated satisfactorily. The highest selectivity observed being ca. 11:1 in favor of the mono substitution product 19 when Na3PO4 was used as the base.
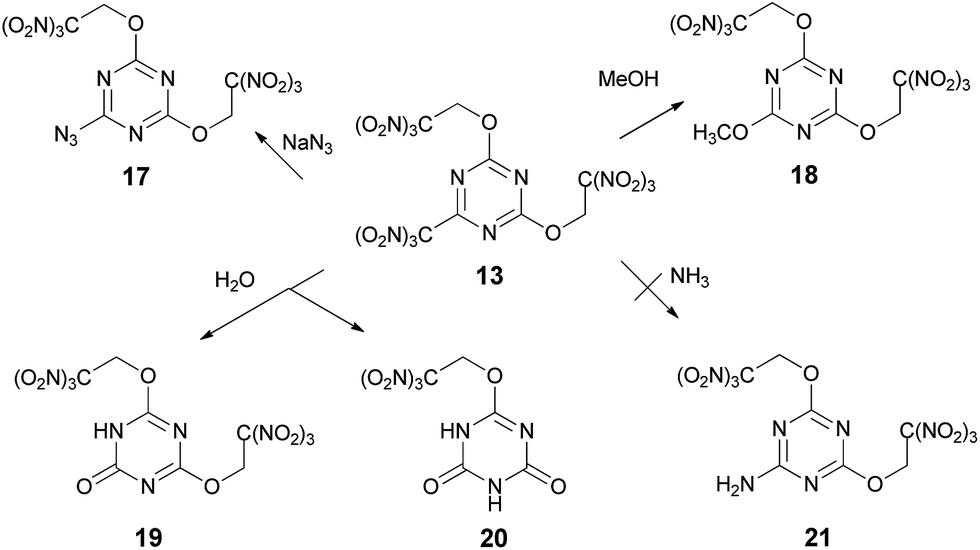 | ||
| Scheme 5 | ||
Unfortunately, reactions of compound 13 with ammonia or various ammonium salts, such as, for example, NH4HCO3 or (NH4)2HPO4, gave intractable tars. None of the product 21 formed by displacement of the trinitromethyl group was isolated. The exact pathway of the decomposition is unclear, but these results demonstrate that reaction of compound 13 with ammonia is unselective.
Since attempts to nucleophilic displacement of the trinitromethyl group were unsuccessful using either ammonia or less reactive ammonium salts, we explored the reaction of compound 16, incorporating three trinitroethoxy groups. Exchange one trinitroethoxy leaving group of compound 16 to amino group now proceeded in 82% yield by treatment with ammonia in DCE to provide required amine 21 (Scheme 6).
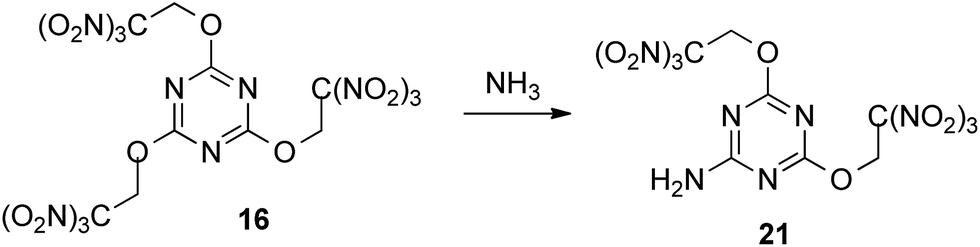 | ||
| Scheme 6 | ||
Compound 16 reacted efficiently with a range of primary and secondary amine nucleophiles, yielding desired products that arise from selective substitution of one leaving group (see the ESI†).
NMR spectroscopy
These trinitroethoxy triazines were fully characterized by NMR, IR, and elemental analysis (see ESI†). Multinuclear NMR spectroscopy proved to be a valuable tool for a characterization of all prepared trinitroethoxy-1,3,5-triazines. In the 1H NMR spectra, the methylene group located between the trinitromethyl carbon and the ether bond was observed as a singlet at ca. δ = 6.4 ppm.In the 13C NMR spectra, the position of resonances of the 1,3,5-triazine ring carbon were in full agreement with literature data.17 A weak signal of the carbon of the trinitromethyl group in the trinitromethyl derivative 13 could be observed at δ = 121–122 ppm. On the other hand, a weak signal of the carbon of the trinitromethyl group in trinitroethoxy moiety was observed at ca. δ = 124 ppm. The structure of compound 16 is symmetric and, correspondingly, three peaks were observed in 13C NMR spectrum.
The 14N NMR spectra of all trinitroethoxy compounds contained a singlet at δ = (−35 ± 3) ppm due to the NO2 group of the trinitroethoxy moiety. In trinitromethyl derivative 13, the nitrogen atoms of C(NO2)3 group signal is found at δ = −22.8 ppm.
The 15N NMR spectrum of compound 17 is shown as an example in Fig. 2. There are three signals (N1, N2, and N3) due to azido group. Also, two peaks (N4 and N5) arising from two nitrogen atoms of the triazine ring, one proximal to the azido group and the second located between the trinitroethoxy moieties, appeared at δ = −172.0 and −181.1 ppm, respectively. The peak (N6) due to the nitrogen atom of the nitro groups is at d = −31.1 ppm.
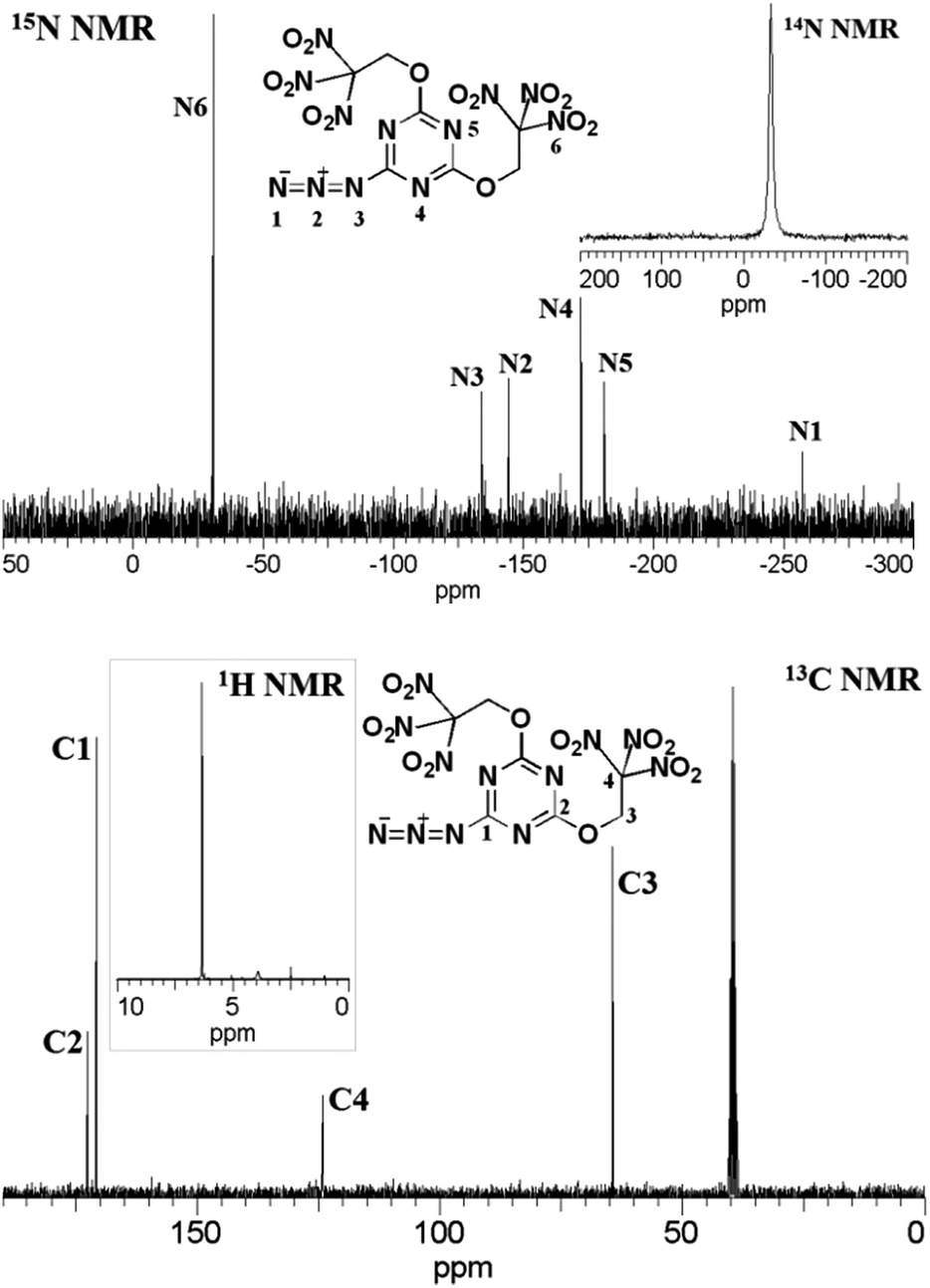 | ||
| Fig. 2 The 1H, 13C, 14N, and 15N NMR spectra of compound 17. | ||
Molecular and crystal structure
Crystals of the compounds 16, 20, and 21 suitable for X-ray diffraction were grown by slow solvent evaporation from CHCl3 solutions at room temperature and normal pressure. The compounds are the first examples of a structural determination of a heteroaryl trinitroethyl ether. Their structures are shown in Fig. 3, while selected geometry characteristics are summarized in Table 1S in ESI.†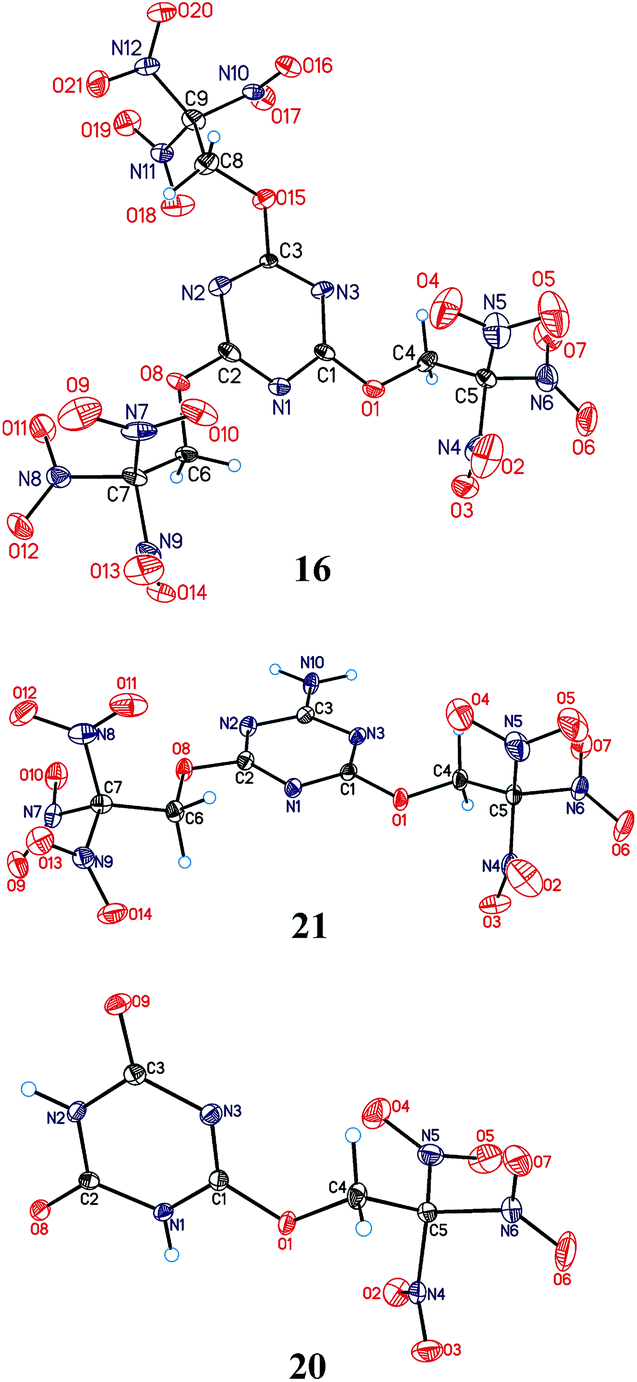 | ||
| Fig. 3 General view of triazines 16, 21, and 20 with the atom numbering scheme. Displacement ellipsoids are drawn at the 50% probability level. For compound 16, the first independent molecule is shown; for compound 21 minor part of the disorder is omitted for clarity. | ||
The geometry and relative orientation of the trinitroethoxy moiety in these compounds are found to be different. Both nearly propeller type orientation and significant deviation from C3 symmetry are observed for the C(NO2)3 groups. Different conformations related to a rotation about O–C bond are also observed. However, nearly constant for all trinitroethoxy moieties appears the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O–CH2 torsion angle being close to 180°. The above-mentioned diversity in the conformation of the trinitroethoxy moiety is an indication of its flexibility and, as we believe, the flexibility allows triazine compounds of this study to form dense crystal structures.
C–O–CH2 torsion angle being close to 180°. The above-mentioned diversity in the conformation of the trinitroethoxy moiety is an indication of its flexibility and, as we believe, the flexibility allows triazine compounds of this study to form dense crystal structures.
For the analysis of the crystal packing we applied combination of the widely used visual inspection of shortened intermolecular contacts (geometrical approach) and approach based on pair intermolecular energies of the molecule with its closest neighbours18 (see details in ESI†).
The triazine 16 contains three independent molecules (A, A′, A′′) in an asymmetric unit cell which have different numbers of closest neighbours and different pattern of intermolecular interactions (Fig.1S, Tables 2S–4S in ESI†). Number of pair interactions between molecules of the same type (A⋯A, A′⋯A′, A′′⋯A′′) is relatively low, and the strongest interactions are observed between symmetrically independent molecules (A′⋯A′′) (Fig. 2S in ESI†). This can be a reason of existence of three independent molecules in the unit cell. Such reasons are usually used for explanation of the co-crystal or solvate formation or the presence of more than one symmetrically independent molecule in the unit cell.2m,19 At the same time, total stabilization energies of A, A′, A′′ (sums of all pair interactions for each molecule) are only slightly different (Tables 2S–4S in ESI†) in spite of completely different molecular environment. This observation outlines crucial role of the trinitroethoxy moiety20 whose flexibility allows such compounds to form dense crystal packing.
Compound 21 contain acidic hydrogen atoms and forms centrosymmetric dimers. The interaction in H-bonded dimer is the strongest one in the crystal structure, and it is much stronger than all other interactions (Table 5S in ESI†). It might be guessed that formation of relatively strong H-bonds would lead to some restrictions of the opportunity of molecules to come closer to each other (densities of ice and water can be served as a classic example: stronger H-bonding in the ice leads to a decrease of its density relative to the liquid state). However, density of compound 21 (1.882 g cm−3) is slightly higher than that of compounds 16 (1.879 g cm−3). We believe that two trinitroethoxy moieties, due to their flexibility, compensate for restrictions caused by H-bonding.
In the crystal of compound 20, each molecule has three H-bonded neighbors (Fig. 4, see also Table 6S in ESI†) and two neighbors connected by short C–H⋯O contacts. Those H-bonds leads to formation of layers parallel to the bc crystallographic plane which are coated with the nitro groups (Fig. 4). Interlayer connection is provided only by nitro⋯nitro interactions being significantly weaker. At the same time, a number of close O⋯O contacts (shorter than 3 Å) between layers are observed, and the fact that packing density of compounds 20 is quite high (1.873 g cm−3, only slightly lower than that of compounds 16 and 21) means that close packing exists not only inside the layers but also between them. It appears that in the case of crystal structure of compounds 20 which is stabilized by both weak peak/hole and strong H-bonded interactions, analysis of crystal packing based on energetic characteristics is insufficient. While energetic criteria work well for explanation of the layer stabilization (93% of the total energy), interlayer interactions are well described by Kitaigorodsky aufbau principle21 that is based on the shape similarity principle and a tendency of molecules to form dense crystal packing.
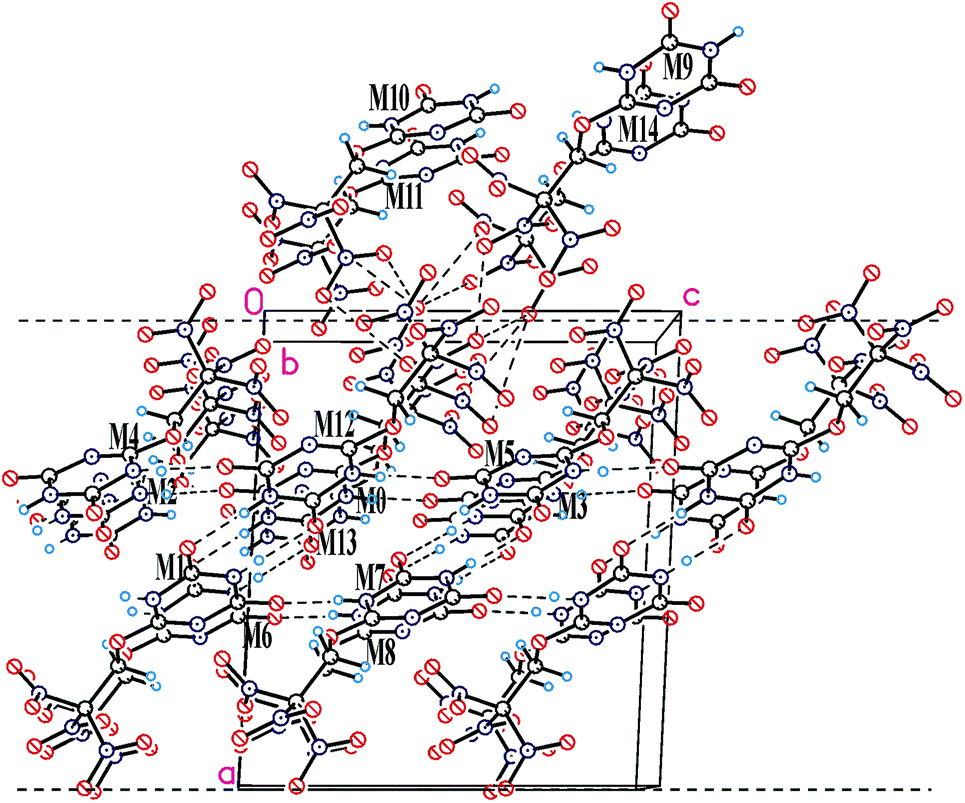 | ||
| Fig. 4 View of the closest environment of the central molecule (M0) of compounds 20 showing H-bonded layer parallel to bc crystallographic plane. The layer thickness is shown by dashed lines, and correspond to the unit cell parameter a (13.2903(13) Å). | ||
Molecular and crystal structures of trinitroethoxy triazines studied here can be compared with their aliphatic analogs (Table 1). For comparison we chose a number of energetic trinitroethoxy compounds with low hydrogen content and high density. Typically, these compounds are esters of one-carbon carboxylic acids; the central fragment bearing trinitroethoxy group is a carbonyl group as a key structural component.
| Entry | Compound | Densitya | Ref. |
|---|---|---|---|
| a Density (g cm−3) recalculated at room temperature (298 K) for easily comparison (see Table 7S in ESI). | |||
| 1 | HOCH2C(NO2)3 | 1.777 | 22 |
| 2 | OC[OCH2C(NO2)3]2 |
1.908 | 23 |
| 3 | (O2N)3CCH2N(NO2)CH2–C(O)–OCH2C(NO2)3 |
1.881 | 24 |
| 4 | (O2N)3CCH2N(NO2)CH2NH–C(O)–OCH2C(NO2)3 |
1.776 | 24 |
| 5 | H2N–C(O)–OCH2C(NO2)3 |
1.799 | 25 |
| 6 | O2NHN–C(O)–OCH2C(NO2)3 |
1.712 | 25 |
| 7 | OC[HN–C(O)–OCH2C(NO2)3]2 |
1.858 | 26 |
| 8 | CH2[OCH2C(NO2)3]2 (TEFO) | 1.813 | 27 |
| 9 | C[OCH2C(NO2)3]4 | 1.905 | 27 |
Both carbonyl and triazine based trinitroethoxy compounds demonstrate similar variety of orientations of the C(NO2)3 group (torsion angles ω and ξ), and different conformations with respect to rotation about O–C bond (torsion angle φ) are observed (see, Fig. 5 and Table 1S in ESI†); for aliphatic molecules from Table 1, the range of φ is 117–167°; the ranges of ω are 39–52°, 73–86° and 153–170°; the ranges of ξ are 20–62° and 114–160°.
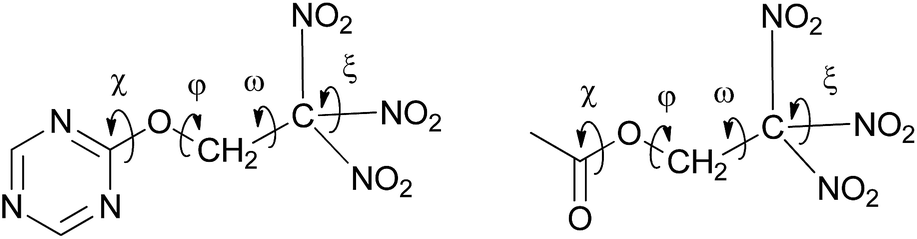 | ||
| Fig. 5 Schematic view of torsion angles which define geometry and relative orientation of the trinitroethyl moiety at the triazine (left) and the carbonyl cores (right). | ||
However, deviation from the planarity (torsion angle χ) of NC–O–C fragment (for triazine core derivatives) somewhat differs from that of OC–O–C fragment (for carbonyl core derivatives). While for the former, deviations of χ does not exceed 9°, for the latter, deviations are more pronounced (for instance, the value of χ in bis(2,2,2-trinitroethyl) carbonate (entry 2, Table 1) is equal to 17.4(2)°), that implies somewhat better flexibility of carbonyl-based trinitroethoxy derivatives. This fact can probably explain nearly isotropic crystal packing of bis(2,2,2-trinitroethyl) carbonate (for packing analysis of this compound see ESI, Fig. 3S and Table 8S†). As a result, its density is higher than that of trinitroethoxy triazines.
As illustrated in Table 1, only in those cases when both substituents at the carbonyl core appears to be trinitroethoxy- or tetranitroethylamino groups then packing density is close to or exceeds 1.9 g cm−3.
It is of importance that our research has shown that variation of substituents at the 1,3,5-triazine core does not lead to significant decrease of packing density that opens an opportunity for further design of energetic triazine materials based on the optimization at the core.
The above results on packing density analysis of triazines show that there are no exact and unambiguous correlation between strength of intermolecular interaction and crystal packing density. It seems that moderate-to-weak interactions formed by trinitroethoxy moiety are responsible for high packing density to a greater extent than strong hydrogen bonds. Such weak interactions (O⋯O, O⋯N, etc.) do not restrict molecular freedom of movement that allows molecules to come closer to each other. This conclusion is supported by a comparison of trinitroethoxy triazines with 2,4,6-triamino-1,3,5-triazine (melamine)28 and 1,3,5-triazinane-2,4,6-trione (cyanuric acid).29 Once all the trinitroethoxy moieties in compound 16 are replaced with the NH2 groups, thus forming melamine, packing density decreases dramatically (1.609 g cm−3 at 100 K for melamine) in spite of the presence of 3-D network stabilized by strong hydrogen bonding. Similar but slightly less pronounced decrease of density is also observed for cyanuric acid (details are given in ESI, Fig. 4S, Table 9S†).
Physical and computational properties
All triazine trinitroethyl ethers are white odorless solids. Physical characteristics and some key properties of these energetic materials are given in Table 2. To assess the potential of the newly prepared trinitroethyl ethers, their energetic characteristics are compared with bis-2,2,2-trinitroethyl formal (TEFO), 2,2,2-trinitroethyl 4,4,4-trinitrobutanoate (O) and PETN. The total nitrogen and oxygen percentages of the new ethers fall in the range of 78–83%. The oxygen excess coefficient (α) is the index of the deficiency or excess of oxygen in a compound required to convert all carbon atoms to CO2, and all hydrogen into H2O. The most oxygen rich compound 13 has the α value of 1.111, which is higher than that of TEFO (α = 1.077). Tris-ether 16 is optimally balanced energetic material (α = 1; ΩCO2 = 0). Compounds 17–19 and 21 possess the α values a bit smaller than 1. However, even for methoxy derivative 18, the α value is equal to 0.77, which is distinctly better than that of conventional plasticizer such as methoxydinitrobenzene (α = 0.294). The experimental densities of ethers 13, 16, 17, 19, and 21 range between 1.8 and 1.9 g cm−3, which is comparable or better than those currently used compounds TEFO, O and PETN.| a Nitrogen content. b Combined nitrogen and oxygen content. c Oxygen balance. For a compound with the molecular formula of CxHyNwOz, ΩCO2 [%] = 1600 [(z − 2x − y/2)/Mw].30 d Oxygen coefficient. For a compound with the molecular formula of CxHyNwOz, α = z/(2x + y/2). A compound with α > 1 is an oxidizer.30 e Density from X-ray analysis. f Melting point. g Decomposition temperature measured at a heating rate of 5 and 10 °C min−1. h Ignition points at constant heating rate (20 °C min−1) and at a constant temperature corresponding to 5 seconds of delay. i Impact sensitivity shown as frequency of explosions at the impact with energy 4.9 J (K-44-II impact machine, set no. 1). j Calculated enthalpy of the formation for solid state. k Detonation velocity at maximal density. l Detonation pressure. m Heat of explosion. n Explosion temperature. o Volume of detonation gases. p Density from pycnometer experiments. | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | 13 | 16 | 17 | 18 | 19 | 21 | TEFO | O | PETN |
| Formula | C8H4N12O20 | C9H6N12O21 | C7H4N12O14 | C8H7N9O15 | C7H5N9O15 | C7H6N10O14 | C5H6N6O14 | C6H6N6O14 | C5H8N4O12 |
| M w | 588.18 | 618.21 | 480.18 | 469.19 | 455.17 | 454.18 | 374.13 | 386.14 | 316.13 |
| N [%]a | 28.58 | 27.19 | 35.00 | 26.87 | 27.70 | 30.84 | 22.46 | 21.76 | 17.72 |
| N + O [%]b | 82.98 | 81.54 | 81.65 | 78.02 | 80.41 | 80.16 | 82.33 | 79.77 | 78.45 |
| Ω CO2 [%]c | 5.4 | 0 | −6.7 | −15.3 | −5.3 | −10.6 | 4.3 | −4.1 | −10.1 |
| α d | 1.111 | 1.0 | 0.875 | 0.77 | 0.909 | 0.824 | 1.077 | 0.933 | 0.857 |
| ρ 20 [g cm−3]e | 1.88p | 1.82 | 1.80p | 1.74p | 1.89p | 1.82 | 1.7 | 1.839 | 1.77 |
| T m f [°C] | 161–162dec | 141–142dec | 115–116 | 58–59 | 170dec | 115–116 | 66–66.5 | 92–92.3 | 141dec |
| Exo. onset/peak DSC (5 °C) [°C]g | 169.5/183.4 | 168.8/184.9 | 169.9/184.5 | 173.7/186.8 | 167.6/181.5 | 148.9/159.0 | 192.1/199.6 | 184.2/191.1 | 162.6/195.8 |
| Exo. onset/peak DSC (10 °C) [°C]g | 182.5/205.6 | 182.8/206.8 | 183.1/207.1 | 183.4/203.3 | 176.6/196.7 | 162.4/180.6 | 200.9/222.4 | 197.4/215.7 | 176.8/207.3 |
| T ign [°C]h | 186 | 201 | 193 | 190 | 190 | 169 | 215 | 207 | 209 |
| T ign(5) [°C]h | 230 | 240 | 236 | 225 | 223 | 208 | 245 | 234 | 233 |
| F [%]i | 36 | 16 | 40 | 12 | 12 | 8 | 16 | 20 | 28 |
| ΔH°f (s) [kJ mol−1]j | −350.4 | −677.5 | −100.9 | −612.1 | −701.3 | −439.9 | −396.1 to 402.1exp31 | −488.5 to 496.3exp32 | −537.1 to 538.7exp33 |
| D [m s−1]k | 8630 | 8460 | 8490 | 8040 | 8330 | 8290 | 8260 | 8400 | 8330 |
| P C–J [GPa]l | 32.8 | 32.2 | 33.1 | 29.0 | 30.0 | 31.1 | 30.1 | 31.0 | 29.8 |
| Q expl [kJ kg−1]m | 6010 | 5552 | 5343 | 5195 | 4815 | 4920 | 6025 | 5790 | 5800 |
| T ex [K]n | 4700 | 4750 | 4650 | 4300 | 4200 | 4200 | 5000 | 5100 | 4450 |
| V eg [l kg−1]o | 680 | 690 | 705 | 715 | 685 | 720 | 730 | 720 | 765 |
The thermal stability of these ethers were investigated by various differential thermal analysis (DTA) measurements (heating rates of 5 °C min−1 and 10 °C min−1). The results of these scans are summarized in Table 2. While most of the compounds showed onset decompositions in the DTA scans at temperatures of ca. 169 °C (at 5 °C min−1) and above, amino compound 21 is the thermally least stable derivative among the investigated trinitroethoxy triazines with a decomposition temperature of ca. 149 °C. The stability level is typical for energetic compounds bearing trinitroethoxy group,34 which is in the same range as observed for PETN.
Upon heating above the decomposition temperature, intense darkening of the trinitroethoxy compounds indicated onset of rapid auto catalyzed reactions leading to spontaneous self-ignition. Special ignition experiments were performed with two standard procedures that previously have been described in detail.35 In the first approach, the ignition points (Tign) were determined at constant heating rate (20 °C min−1). In the second approach, fire test was achieved at a constant temperature (Tign(5)) corresponding to 5 seconds of delay time of the ignition. These ignition points for new compounds and analogs are listed in Table 2. Two rows of the table show how the ignition points change from compound to compound during measurements with both procedures. Essentially, the trends of temperatures are very similar in each row, when the temperature Tign(5) is more than the temperature Tign to 35–40 °C.
The enthalpy of formation (ΔH°f) is a very important parameter in evaluating the performance of energetic materials. This characteristic can be calculated for energetic 1,3,5-triazines by various quantum chemical methods.36 However, recently it has been shown that for trinitroethoxy compounds2c,37 a simple prediction of ΔH°f can be performed with a good accuracy by using the additive method based on the values of the group contribution and the intramolecular interaction.38 The enthalpies of formation for new compounds were calculated and are summarized in Table 2. For comparison, we calculated the heats of formation for compounds TEFO and O and PETN, for which experimental data are available. Naturally, the azido compound 17 has the highest enthalpies of formation for this series of energetic compounds. The calculated ΔH°f values vary between −1.541 (for 19) to −0.210 kJ g−1 (for 17), which are much higher than that of PETN (−1.704 kJ g−1).
Using the calculated enthalpies of formation values together with the experimental density measurements, the performance of these trinitroethoxy triazines were estimated using the computational methods developed by Smirnov et al.39 The results of these studies can be found in Table 2. With the exception of compound 18, all of them have similar or higher detonation pressures and velocities than compound TEFO. Thus, the detonation pressures (PC–J) for the triazines fall in the range of about 30 to 33 GPa, and the detonation velocities (D) are distributed from 8290 (for 21, comparable to PETN, PC–J = 29.8 GPa, D = 8340) to 8630 ms−1 (for 13, comparable to RDX, PC–J = 33.8 GPa, D = 8750). It should be noted that compound 13, an oxidant with maximal oxygen coefficient (α = 1.111), exhibits better performance than compound TEFO (α = 1.077).
The impact sensitivities of the trinitroethoxy triazines were determined using a K-44-2 impact machine (set. no. 1).40 The determined sensitivities are summarized in Table 2. Compounds 13 and 17 are more sensitive to impact than PETN run on the same instrument under the same conditions (see Table 2). Other trinitroethoxy-containing compounds are approximately two times less sensitive than PETN. The impact sensitivity of compound 16 and TEFO are similar. In general, however, these trinitroethoxy compounds are relatively sensitive.
Propellant evaluation
The objective of this section is theoretically examined the use of triazine compounds of this study as ingredients for solid composite rocket propellants (SCRP) and gas-generating compositions for airbag inflators.Theoretical performance calculations were performed using the TERRA code.41 Here for model SCRP the specific impulse (Isp) was used as the performance parameter of merit, calculated assuming a chamber pressure of 4.04 MPa and ideal sea level equilibrium expansion. Estimated model propellants close to real, where one of a common ingredient was partially or completely replaced by a new compound.
A typical aluminized SCRP, ammonium perchlorate (AP, 70.5%)–hydrocarbon binder (HCB, 9.5%)–aluminum (Al, 20%),42 was selected as the backbone. Chlorine free replacement for AP, which will yield similar or superior performance, is very desirable.43 The combustion parameters of the formulations, where a new compound is used as a co-oxidizer together with AP, were calculated. The resulting performance contours are shown in Fig. 6.
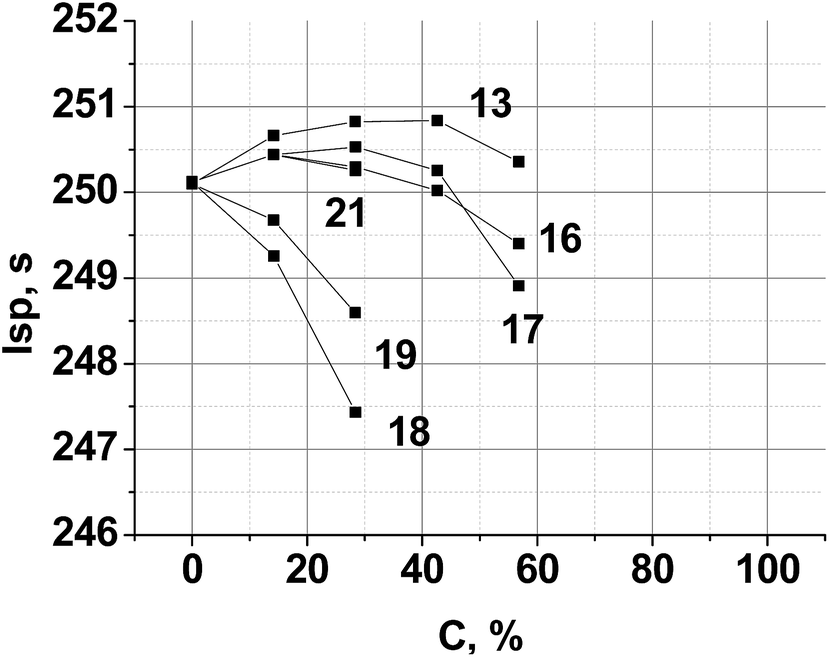 | ||
| Fig. 6 Calculated specific impulse of (AP + new compound)–Al(20%)–HCB(9.5%) propellants over various new compound loadings. Here, C is the percentage of a new compound in the mixture with AP. HCB is hydrocarbon binder (C73.17H120.9, ΔH°f = −393 kJ kg−1, ρ = 0.91 g cm−3).44 | ||
Because of the highest α value (1.111) and the higher enthalpy of formation ΔH°f (−596 kJ kg−1) (see, Table 2), compound 13 is the best AP replacement in SCRP. The inclusion of compounds 16, 17 and 21 is most preferable over the range 20–40% surpass an Isp of 250 s, near that of existing AP-based SCRP. Fig. 6 also shows that with compound 18 and 19 as co-oxidizer to AP, Isp was decreased drastically because of low ΔH°f values of these triazines (see, Table 2).
Next, this study has been extended to consider similar formulations, where HCB was replaced by an energetic binder AB44 (this binder contains nitro groups, C18.96H34.64N19.16O29.32, ΔH°f = −757 kJ kg−1, d = 1.49 g cm−3).
As shown by the data in Fig. 7, at every new compounds loading level from 1 to 100%, superior performance is achieved by the AB containing solid propellant formulations. Indeed, the oxygen contents of the backbone Al–AB–PA formulation is too high and it is needed to decrease it. In this case compounds 13, 16 and 19 have the highest α values and are not very effective as AP replacement. Nevertheless, these compounds as the co-oxidizers increase the Isp values. The majority of the improved performance can be attributed to the fact that increasing new compound concentration improves CO2-balance of the propellant formulation. Formulations with compound 17 appear to exhibit the largest increase in the specific impulse compared to their PA and other triazine-based formulation counterparts. The increase in performance seen in this case comes from several properties directly attributed to the compound 17, including its rather high ΔH°f value and relatively low α.
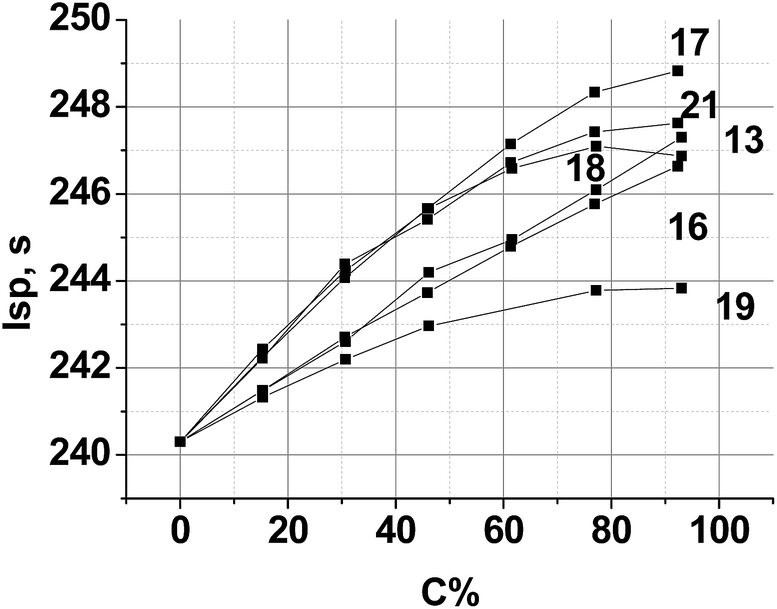 | ||
| Fig. 7 Calculated specific impulse Isp of (AP + new compound)–Al(20%)–AB(14–15%) propellants over various new compound loadings. Here, C is the percentage of a new compound in the mixture with AP. | ||
Since triazine compounds of this study are flexible molecules with relatively low melting points, it is possible to consider them as prospective ingredients of a plasticizer,45 in which they partially replace commonly used nitroglycerin (NG), e.g. in the known formulation with 25% aluminum hydride, 50% ammonium dinitramide (AND),46 5% polymethylvinyltetrazole (PMVT), and 20% NG.47 As indicated in Fig. 8a, the calculated specific impulse of these triazine-based propellant formulations lies between 273 and 277 s. At every new compounds replacement, a slight decrease in Isp is achieved by the propellant formulations.
The least negative effect was observed in the case when NG (α = 1.06, ΔH°f = −1630 kJ kg−1, hydrogen content 2.2%) was replaced by compound 13. The compound has practically the same α value as NG, and considerably higher ΔH°f. However too low hydrogen content (0.7%) does not allow getting benefit from a high ΔH°f value of compound 13. The use of compound 19 is the least effective. Its ΔH°f is close to ΔH°f of NG, but both α and hydrogen content are lower.
The data in Fig. 8a clearly illustrate that at 50% replacement of NG by compounds 13, 16, 17, 18, or 21 the value Isp decrease is less than 1.5 s. If this mixture (NG/new compound = 1/1) is less sensitive, than neat NG (there are enough thorough presuppositions for this affirmation) such replacement may be perspective.
 | ||
| Fig. 8 (a) Calculated specific impulse Isp of ADN(50%)–AlH3(25%)–PMVT(5%)–(NG + new compound) (20%) propellants over various new compound loadings. Here, C is the percentage of a new compound in the mixture with NG. (b) Ief(3) characterized the ballistic effectiveness of the propellants utilized in the third stage of rocket complex, Ief(3) = Isp + 25 × (ρ − 1.7).48 It is a relative effectiveness of propellants with different densities. | ||
All triazine compounds of this study (see Table 2) are more dense than NG (1.59 g cm−3) providing heightened density of propellants containing them. The ballistic effectiveness of these propellant formulations slightly lower than formulations with neat NG (Fig. 8b).
Formulating gas generant pyrotechnic composition (GGC) for any particular application requires balancing of a number of parameters, such as gas volume per weight of generant composition, burn rate, combustion temperature, etc.49 Gas generant propellants commonly utilized in the inflation of automotive inflatable restraint airbag cushions have previously most typically been based on sodium azide as the fuel, and include any transition metal oxide (typically, CuO or Fe2O3) as an oxidizer. Such azide-based propellants, upon initiation, normally produce harmless nitrogen gas. However, these metal-containing compositions also generate considerable amounts of condensed-phase residue that must be removed from the products prior to entering the airbag. Other disadvantages of sodium azide are toxicity and particularly instability in the presence of metallic impurities.50
Alternatives to azides which have been proposed include high nitrogen compounds, such as tetrazole and triazole derivatives.51 When combined with an oxidizer, and formed into a gas generant composition, this non-azide propellants compose of carbon, hydrogen, nitrogen and oxygen atoms typically yield greater amounts of gas upon combustion. Most oxidizers commonly employed in azole-based gas generant compositions are metal salts of oxygen-rich anions (for example, nitrates, chlorates or perchlorates). Unfortunately, upon combustion, the metallic components of such oxidizers typically end up as a solid and thus reduce the relative gas yield realizable therefrom. Consequently, the amount of such oxidizers and a particular formulation typically affects the gas output or yield from the formulation. A problem encountered with gas generant compositions which utilize tetrazoles or triazoles as fuel is the production of undesirable gases, such as CO, NOx, NH3, and HCN. However, the automotive industry is becoming increasingly sensitive to undesirable combustion products (gases and solid residue) and is imposing increasingly stricter standards. There is an ongoing need for further improved, safe and effective alternative gas generant formulations employed non-azide metal-free fuels and oxidizers, that upon combustion produce a relatively small amount of solids and a relatively abundant amount of gas.
If functional groups with active oxygen are incorporated into a fuel component less of an oxidizer may be required and the gas output of the propellant can be increased.52
In fact, ingredients for gas generant pyrotechnic compositions that are favored because of their propensity to produce all or mostly gas are triazine compounds of this study. When compounds 17–19 and 21 (α < 1) combined with an oxidizer, and formed into a gas generant composition, this fuel contributes to greater amounts of gas upon combustion of the composition (Table 3). A metal-free oxidizer may be selected from nitrate salts of amines or substituted amines; from an availability and cost standpoint, ammonium nitrate (AN)53 is presently the oxidizer of choice. The purpose of the oxidizer is to provide the oxygen necessary to oxidize the fuel (α = 1; to stoichiometrically yield carbon dioxide, nitrogen, and water); for example, NH4NO3 oxidizes compound 21 according to the following equation:
| C7H6N10O14 + 3NH4NO3 → 7CO2 + 8N2 + 9H2O |
| Entry | Oxidizer (wt%) | Fuel (wt%) | Gas volumea, mol kg−1 | T comb, K |
|---|---|---|---|---|
| a CO2 and N2 only, because namely these gases volume characterizes the capacity of this kind of GGC. | ||||
| 1 | NH4NO3 (75.0) | 17 (25.0) | 23.4 | 3140 |
| 2 | NH4NO3 (56.6) | 18 (43.4) | 20.5 | 3050 |
| 3 | NH4NO3 (79.1) | 19 (20.9) | 22.6 | 3027 |
| 4 | NH4NO3 (65.4) | 21 (34.6) | 21.6 | 2963 |
| 5 | NH4NO3 (82.8) | O (17.2) | 21.6 | 3240 |
| 6 | NH4NO3 (66.4) | PETN (33.6) | 18.9 | 3048 |
| 7 | 16 (100) | 24.3 | 3336 | |
| 8 | 13 (76.6) | Oxalic acid (23.4) | 23.4 | 3467 |
| 9 | TEFO (80.6) | Oxalic acid (19.4) | 21.6 | 3453 |
The gas yield is ca. 0.02 moles of gas per 1 gram of the formulation.
Compound 16 (α = 1) is mono-propellant; intra-molecular redox reaction produces an estimated amount of gases.
As an oxygen-rich energetic material, compound 13 (α = 1.11) appears to hold potential as an oxidizer. Proposed fuels for use in conjunction with oxidizer 13 include the organic compounds containing carboxylic acid groups or carboxylic acid salt groups. A currently preferred fuel is oxalic acid. The theoretical predictions of this composition indicate that there is a considerable increase in combustion temperature, comparable to TEFO–oxalic acid system (Table 3).
Moreover, combustion temperatures (Tcomb) of all stoichiometric formulations are considerably higher than it is required (lower than 2500 K)54 for ensure low level of CO and nitrous gases (these gases may be controlled by formulating to burn at sufficiently low temperatures where their production are minimized). Anyway on the average combustion temperatures of triazine-based stoichiometric formulations are a bit lower than for formulations based on TEFO, O and PETN (Table 3).
Conclusions
A number of novel energetic trinitroethyl ethers, 2,4-bis(2,2,2-trinitroethoxy)-6-R-1,3,5-triazines, have been prepared. These compounds represent the first hetaryltrinitroethyl ethers. Key 2,4-bis(2,2,2-trinitroethoxy)-6-trinitrometyl-1,3,5-triazine 13 can be readily synthesized in one-pot multicomponent processes from commercially available 2,4,6-trichloro-1,3,5-triazine. The compound 13 can act as functional core scaffolds and react further with nucleophiles to give products arising from substitution of trinitromethyl group, demonstrating the potential of the triazine as a scaffold for the synthesis of previously inaccessible hetaryltrinitroethyl ethers. In particular, we have synthesized 1,3,5-triazine persubstituted with trinitroethoxy moieties, 2,4,6-tris(2,2,2-trinitroethoxy)-1,3,5-triazine 16. For the first time, heteroaryltrinitroethyl ethers were fully characterized, including X-ray structure determinations. Analysis of their crystal packing based on combination of geometrical and energetic approaches has reviled that nitro groups participate in nearly all intermolecular interactions. Such interactions are of moderate-to-weak force and are responsible for high density of the crystal packing of trinitroethoxy triazines allowing molecules to come closer to each other.All new ethers exhibited reasonable physical properties, such as a high density, good oxygen balance and good thermal stability. Calculations indicate that their energetic performance exceeds that of TEFO, and PETN. Concomitantly, this compound shows acceptable impact sensitivities and ignition points. Low melting points make them suitable candidates as a melt-castable energetic plasticizer in formulations of energetic binders. Since some of the new trinitroethyl ethers have good oxygen and nitrogen content, the calculated specific impulses confirm that these compounds can be considered as suitable replacement for common ingredients in solid propellants. Various combinations of the new trinitroethyl ethers and metal-free oxidizers/fuels have been proposed for use in gas generant compositions that upon combustion produce relatively high gas outputs and show better characteristics than TEFO, O and PETN.
Thus, this general approach has special relevance to the development of new environmentally friendly ingredients for the energetic material industries and particularly in the variety propellant discovery arena, in which nitrogen or oxygen-rich polyfunctional heterocyclic derivatives are playing an increasingly important role.
Experimental
Caution! All prepared compounds are highly energetic materials and sensitive to various stimuli. Although none of the compounds described herein has exploded or detonated in the course of this research, safety precautions, such as face shields, a leather apron, gloves, and hearing protection should be employed. These compounds should only be prepared by an experienced synthetic chemist and should be handled with great care.General
All the reagents were of analytical grade, purchased from commercial sources, and used as received. Sodium trinitromethanide,55 potassium trinitromethanide,56 and trinitroethanol57 were synthesized by using previously reported methods. Infrared spectra were determined in KBr pellets on a Perkin-Elmer Model 577 spectrometer. Mass-spectra were recorded on a Varian MAT-311 A instrument. The 1H, 13C, 15N, and 14N NMR spectra were recorded on a Bruker AM-300 instrument at 300.13, 75.47, 50.7, and 21.68 MHz, respectively. The chemical shift values (δ) are expressed relative to the chemical shift of the [D]solvent or to external standard (nitromethane for 14N and 15N NMR) without correction. Analytical TLC was performed using commercially pre-coated silica gel plates (Silufol UV254), and visualization was effected with short-wavelength UV-light. Melting points were determined on Gallenkamp melting point apparatus and they are uncorrected. The decomposition points were recorded on a thermogravimetric analyzer (TGA) at a scan rate of 5 °C min−1 or 10 °C min−1. Elemental analyses were obtained by using a CHNS/O Analyzer 2400 (Perkin-Elmer instruments Series II).X-ray crystallography
X-ray experiments were carried out using SMART APEX2 CCD (λ(Mo-Kα) = 0.71073 Å, graphite monochromator, ω-scans) at 100 K and 298 K. Experimental details are given in Tables 10S, 11S in ESI.†2,4-Bis(2,2,2-trinitroethoxy)-6-(trinitromethyl)-1,3,5-triazine (13)
To a solution of trinitroethanol (4.53 g, 25 mmol) in absolute acetone (12.5 mL) were added trichlorotriazine (1.84 g, 10 mmol) and potassium trinitromethanide (7.56 g, 40 mmol). The resulting mixture was stirred at room temperature for 44–48 h (TLC monitoring). The mixture was filtered and then concentrated (50%) in vacuo. The filtrate was diluted by water (70 mL) to give a suspension that was neutralized to pH 7 with NaHCO3. The precipitate was filtered off, washed with water (4 × 20 mL), and dried to give a bright yellow solid. Recrystallization from DCE gave a colorless solid 13 (1.88 g, 32%), mp 161–162 °C. 1H NMR (acetone-d6): δ = 6.45 ppm (s, CH2). 13C NMR (acetone-d6): δ = 65.4 (CH2), 123.5 (bs, C(NO2)3), 164.5 (NC–C(NO2)3), 171.4 ppm (NC–O). 14N NMR (acetone-d6): δ = −22.8, −34.7 ppm. IR: ν = 3022, 2968, 2889, 1641, 1603, 1591, 1541, 1489, 1437, 1417, 1373, 1340, 1288, 1176, 1132, 1088, 993, 879, 858, 833, 804, 783, 760, 660, 644, 577, 546 cm−1. Elemental analysis calcd (%) for C8H4N12O20 (588.18): C 16.34, H 0.69, N 28.58; found: C 16.31, H 0.79, N 28.64.
2,4,6-Tris(trinitroethoxy)-1,3,5-triazine (16)
To a solution of trinitroethanol (7.2 g, 40 mmol) in absolute acetone (25 mL) were added compound 13 (4.7 g, 8 mmol) and potassium trinitromethanide (1.89 g, 10 mmol). The resulting mixture was stirred at room temperature for 10 d (TLC monitoring). The mixture was filtered and then concentrated (50%) in vacuo. The filtrate was diluted by water (70 mL) to give a suspension that was neutralized to pH 7 with NaHCO3. The precipitate was filtered off, washed with water (4 × 20 mL), and dried to give a bright yellow solid. Recrystallization from CCl4 gave a colorless solid (3.16 g, 64%), mp 141–142 °C (dec.). 1H NMR (acetone-d6): δ = 5.73 ppm (s, CH2). 13C NMR (acetone-d6): δ = 64.6 (OCH2), 122.1 (bs, C(NO2)3), 171.6 ppm (NC–OCH2). 14N NMR (acetone-d6): δ = −36.1 ppm. IR: ν = 3014, 2968, 2895, 1713, 1595, 1497, 1446, 1412, 1369, 1331, 1300, 1155, 1088, 879, 852, 804, 783, 652, 542 cm−1. Elemental analysis calcd (%) for C9H6N12O21 (618.21): C 17.49, H 0.98, N 27.19; found: C 17.44, H 1.03, N 27.17.
2-Azido-4,6-bis(2,2,2-trinitroethoxy)-1,3,5-triazine (17)
A slurry of NaN3 (0.078 g, 1.2 mmol) and compound 13 (0.59 g, 1 mmol) in absolute acetonitrile (10 mL) was stirred at 0–3 °C for 3 h. The mixture was filtered and then concentrated (50%) in vacuo. The filtrate was diluted by water (30 mL) to give a suspension. The precipitate was filtered off, washed with water (4 × 20 mL), and dried to give a bright yellow solid. Recrystallization from isopropanol gave a colorless solid (0.4 g, 84%), mp 115–116 °C. 1H NMR (DMSO-d6): δ = 6.34 ppm (s, CH2). 13C NMR (DMSO-d6): δ = 64.3 (CH2), 124.1 (bs, C(NO2)3), 170.7 (NC–O), 172.6 ppm (NC–N3). 14N NMR (DMSO-d6): δ = −34.2 ppm. IR: ν = 3009, 2955, 2893, 2175, 2154, 2135, 1597, 1551, 1477, 1437, 1412, 1396, 1365, 1304, 1265, 1190, 1144, 1084, 879, 856, 814, 804, 783, 662, 552 cm−1. Elemental analysis calcd (%) for C7H4N12O14 (480.18): C 17.51, H 0.84, N 35.00; found: C 17.54, H 0.87, N 35.06.
2,4-Bis(2,2,2-trinitroethoxy)-6-methoxy-1,3,5-triazine (18)
To a solution of compound 13 (0.4 g, 0.68 mmol) in a mixture of methanol (0.04 mL, 1 mmol) and acetone (13 mL) was added at room temperature potassium carbonate (0.19 g, 1.36 mmol) by small portions during 1 h. The resulting mixture was stirred at room temperature for ca. 6 h (TLC monitoring). The mixture was filtered, concentrated (to 1/3) in vacuo and diluted by water (30 mL). The precipitate was filtered off, washed with water (4 × 20 mL), and dried to give a bright yellow solid. Recrystallization from CCl4 gave a colorless solid (0.19 g, 60%), mp 58–59 °C. 1H NMR (DMSO-d6): δ = 6.29 (4H, s, CH2), 4.04 ppm (3H, s, CH3). 13C NMR (DMSO-d6): δ = 56.4 (CH3), 64.0 (CH2), 124.2 (bs, C(NO2)3), 171.0 (NC–OCH2), 173.3 ppm (NC–OCH3). 14N NMR (DMSO-d6): δ = −34.2 ppm (NO2). IR: ν = 3030, 3002, 2952, 2887, 1598, 1567, 1519, 1461, 1432, 1407, 1492, 1369, 1334, 1292, 1198, 1147, 1117, 1088, 941, 878, 856, 819, 802, 785, 760 cm−1. Elemental analysis calcd (%) for C8H7N9O15 (469.19): C 20.48, H 1.50, N 26.87; found: C 20.52, H 1.61, N 26.83.
4,6-Bis(2,2,2-trinitroethoxy)-1,3,5-triazine-2-one (19A and 19B) and 6-(2,2,2-trinitroethoxy)-1,3,5-triazine-2,4(1H,3H)-dione (20)
A mixture of Na3PO4 (0.2 g, 1.28 mmol) and compound 13 (0.5 g, 0.85 mmol) in 94% aqueous acetone (5.5 mL) was stirred at room temperature for 5–6 h. The mixture was filtered and then concentrated (50%) in vacuo. The filtrate was diluted by a solution NaHCO3 (0.084 g, 1 mmol) in water (20 mL) to give a suspension. The precipitate (compound 20) was filtered off and washed with acetone (5 mL), and the filtrate was acidified to pH 3 with 1 N HCl. The precipitate was filtered off and dried to give compound 19 (0.32 g, 82%), as a mixture of tautomers19A and 19B, mp 170 °C (dec.). 1H NMR (DMSO-d6): δ = 12.79 (1H, bs, NH), 11.34 (1H, s, NH), 6.23 (4H, s, OCH2), 6.17 (2H, s, OCH2), 5.05 ppm (2H, s, OCH2). 13C NMR (DMSO-d6): δ = 170.4, 170.3, 166.8, 159.9, 158.5, 124.0, 123.8, 63.9, 63.5, 62.2 ppm. IR: ν = 3182, 3084, 3022, 2964, 2810, 1749, 1734, 1689, 1610, 1579, 1525, 1441, 1423, 1389, 1300, 1286, 1128, 1095, 957, 852, 841, 802, 783 cm−1. Elemental analysis calcd (%) for C7H5N9O15 (455.17): C 18.47, H 1.11, N 27.70; found: C 18.41, H 1.15, N 27.72.Compound 20 was recrystallized from aqueous AcOH to give a colorless solid (0.017 g, 7%), mp 180 °C (decomp.). IR: ν = 3320, 3230, 3150 (NH); 1797, 1747 (CO) cm−1. Elemental analysis calcd (%) for C5H4N6O9 (292.12): C 20.56, H 1.38, N 28.77; found: C 20.60, H 1.43, N 28.74.
2-Amino-4,6-bis(2,2,2-trinitroethoxy)-1,3,5-triazine (21)
Compound 16 (1 g, 1.62 mmol) was dissolved in DCE (10 mL) and NH3 was bubbled through the solution for 20–25 min (TLC monitoring). A solid was filtered off, the filtrate was evaporated under reduced pressure, and the residue was washed with cold water (2 × 40 mL) and heat water (60 °C, 2 × 40 mL). Recrystallization from CCl4 gave compound 21 (0.61 g, 82%) as a white solid, mp 115–116 °C. 1H NMR (DMSO-d6): δ = 6.13 (s, 4H, OCH2), 8.13 ppm (s, 2H, NH2); 13C NMR (DMSO-d6): δ = 63.8 (OCH2), 124.9 (C(NO2)3), 169.1 (C–NH2), 170.4 ppm (C–OCH2). 14N NMR (DMSO-d6): δ = −35.7 ppm (NO2). 15N NMR (DMSO-d6): δ = −30.4 (NO2), −191.2, −196.2 (triazine), −280.4 ppm (NH2). IR: ν = 3493, 3307, 3196, 3149, 3008, 2962, 2887, 1639, 1585, 1556, 1448, 1406, 1369, 1329, 1299, 1142, 1084, 1045, 879, 856, 804, 783 cm−1. Elemental analysis calcd (%) for C7H6N10O14 (454.18): C 18.51, H 1.33, N 30.84; found: C 18.47; H 1.41; N 30.89.Acknowledgements
Financial support of this work by the Ministry of Education and Science of Russia within the scope on the Project Section of the State task for Samara State Technical University (project No. 4.813.2014/K), and the Presidium of the Russian Academy of Sciences (the program OKhNM-04) are gratefully acknowledged. KYS is thankful to the Russian Scientific Foundation (14-13-00884) for financial support for structural investigations.Notes and references
- (a) V. A. Ostrovskii, M. S. Pevzner, T. P. Kofman, M. B. Shcherbinin and I. V. Tselinskii, Targets Heterocycl. Syst., 1999, 3, 467 CAS; (b) P. F. Pagoria, G. S. Lee, A. R. Mitchell and R. D. Schmidt, Thermochim. Acta, 2002, 384, 187 CrossRef CAS; (c) M. B. Talawar, R. Sivabalan, S. N. Asthana and H. Singh, Combust., Explos. Shock Waves, 2005, 41, 264 CrossRef; (d) High Energy Density Materials, ed. T. M. Klapotke, Springer, Berlin, 2007, vol. 125, Springer-Verlag textbook series on Structure and Bonding Search PubMed; (e) J. P. Agrawal and R. D. Hodgson, Organic Chemistry of Explosives, John Wiley & Sons, Ltd., 2007 Search PubMed; (f) L. Larina and V. Lopyrev, Nitroazoles: Synthesis, Structure and Applications. Springer, LLC, 2009 Search PubMed; (g) V. I. Pepekin, Russ. J. Phys. Chem. B, 2010, 4, 954 CrossRef; (h) V. I. Pepekin, Russ. J. Phys. Chem. B, 2011, 5, 41 CrossRef CAS.
- (a) L. Liang, D. Cao, J. Song, H. Huang, K. Wang, C. Bian, X. Dong and Z. Zhou, J. Mater. Chem. A, 2013, 1, 8857 RSC; (b) V. V. Parakhin and O. A. Luk'yanov, Russ. Chem. Bull., 2013, 62, 2007 CrossRef CAS; (c) A. B. Sheremetev, N. S. Aleksandrova, N. V. Palysaeva, M. I. Struchkova, V. A. Tartakovsky and K. Y. Suponitsky, Chem.–Eur. J., 2013, 19, 12446 CrossRef CAS PubMed; (d) D. Chand, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2013, 1, 15383 RSC; (e) N. V. Palysaeva, K. P. Kupman, M. I. Struchkova, I. L. Dalinger, A. V. Kormanov, N. S. Aleksandrova, V. M. Chernyshev, D. F. Pyreu, K. Yu. Suponitsky and A. B. Sheremetev, Org. Lett., 2014, 16, 406 CrossRef CAS PubMed; (f) A. A. Voronin, V. P. Zelenov, A. M. Churakov, Y. A. Strelenko, I. V. Fedyanin and V. A. Tartakovsky, Tetrahedron, 2014, 70, 3018 CrossRef CAS; (g) M. A. Kettner and T. M. Klapotke, Chem. Commun., 2014, 50, 2268 RSC; (h) L. Zhai, X. Fan, B. Wang, F. Bi, Y. Li and Y. Zhu, RSC Adv., 2015, 5, 57833 RSC; (i) V. Forquet, C. M. Sabate, G. Jacob, Y. Guelou, H. Delalu and C. Darwich, Chem.–Asian J., 2015, 10, 1668 CrossRef CAS PubMed; (j) X. Yin, J.-T. Wu, X. Jin, C.-X. Xu, P. He, T. Li, K. Wang, J. Qin and J.-G. Zhang, RSC Adv., 2015, 5, 60006 Search PubMed; (k) D. E. Chavez, T. W. Myers, J. M. Veauthier, M. T. Greenfield, R. J. Scharff and D. A. Parrish, Synlett, 2015, 26, 2029 CrossRef CAS; (l) R. Haiges and K. O. Christe, Dalton Trans., 2015, 44, 10166 RSC; (m) I. L. Dalinger, I. A. Vatsadze, T. K. Shkineva, A. V. Kormanov, M. I. Struchkova, K. Yu. Suponitsky, A. A. Bragin, K. A. Monogarov, V. P. Sinditskii and A. B. Sheremetev, Chem.–Asian J., 2015, 10, 1987 CrossRef CAS PubMed; (n) I. L. Dalinger, A. K. Shakhnes, K. A. Monogarov, K. Yu. Suponitsky and A. B. Sheremetev, Mendeleev Commun., 2015, 25, 429 CrossRef CAS.
- (a) H. Ritter and H. H. Licht, Propellants, Explos., Pyrotech., 1993, 18, 81 CrossRef CAS; (b) A. V. Shastin, T. I. Godovikova and B. L. Korsunskii, Chem. Heterocycl. Compd., 1999, 35, 75 CrossRef CAS; (c) P. N. Simoes, L. M. Pedroso, A. M. M. Beja, M. R. Silva, E. MacLean and A. A. Portugal, J. Phys. Chem. A, 2007, 111, 150 CrossRef CAS PubMed; (d) A. A. Gidaspov, New Trends in Research of Energetic Materials, Proceedings of the Seminar, 11th, Pardubice, Czech Republic, 2008, p. 540 Search PubMed; (e) Y. Huang, Y. Zhang and J. M. Shreeve, Chem.–Eur. J., 2011, 17, 1538 CrossRef CAS PubMed; (f) F. Wang, H. Du, H. Liu and X. Gong, Chem.–Asian J., 2012, 7, 2577 CrossRef CAS PubMed.
- (a) M. H. Huynh, M. A. Hiskey, E. L. Hartline, D. P. Montoya and R. Gilardi, Angew. Chem., Int. Ed., 2004, 43, 4924 CrossRef CAS PubMed; (b) M. H. Huynh, M. Hiskey, C. Pollard, D. Montoya, E. Hartline and R. Gilardi, J. Energ. Mater., 2004, 22, 217 CrossRef CAS; (c) A. V. Shastin, T. I. Godovikova and B. L. Korsunskii, Russ. Chem. Bull., 2011, 60, 1220 CrossRef CAS; (d) G. V. Malkov, A. V. Shastin, Y. I. Estrin, E. R. Badamshina and Y. M. Mikhailov, Russ. Chem. Bull., 2011, 60, 1940 CrossRef CAS; (e) T. Musil, R. Matyas, A. Lycka and A. Ruzicka, Propellants, Explos., Pyrotech., 2012, 37, 275 CrossRef CAS; (f) Q.-L. Yan, T. Musil, S. Zeman, R. Matyas, X.-B. Shi, M. Vlcek and V. Pelikan, Thermochim. Acta, 2015, 604, 106 CrossRef CAS.
- (a) I. V. Ovchinnikov, A. S. Kulikov, M. A. Epishina, N. N. Makhova and V. A. Tartakovsky, Russ. Chem. Bull., 2005, 54, 1346 CrossRef CAS; (b) S. Li, W. Zhang, Y. Wang, X. Zhao, L. Zhang and S. Pang, J. Energ. Mater., 2014, 32, S33 CrossRef.
- (a) A. V. Shastin, T. I. Godovikova, S. P. Golova, V. S. Kuz'min, L. I. Khmel'nitskii and B. L. Korsunskii, Mendeleev Commun., 1995, 17 CrossRef CAS; (b) A. V. Shastin, T. I. Godovikova and B. L. Korsunskii, Chem. Heterocycl. Compd., 2003, 39, 354 CrossRef CAS; (c) V. V. Bakharev, A. A. Gidaspov and I. V. Ul'yankina, New Trends in Research of Energetic Materials, Proceedings of the Seminar, 13th, Pardubice, Czech Republic, 2010, p. 404 Search PubMed; (d) A. V. Shastin, T. I. Godovikova and B. L. Korsunskii, Russ. Chem. Bull., 2011, 60, 1220 CrossRef CAS; (e) O. V. Golovina, V. V. Bakharev, E. V. Golovin, V. E. Parfenov and P. A. Slepukhin, Tetrahedron Lett., 2013, 54, 3858 CrossRef CAS; (f) A. A. Gidaspov, V. V. Bakharev, O. V. Golovina and E. V. Selezneva, New Trends in Research of Energetic Materials, Proceedings of the Seminar, 17th, Pardubice, Czech Republic, 2014, p. 653 Search PubMed.
- (a) M. H. V. Huynh, M. A. Hiskey, C. J. Pollard, D. P. Montoya, E. L. Hartline and R. Gilardi, J. Energ. Mater., 2004, 22, 217 CrossRef CAS; (b) V. N. Kizhnyaev, F. A. Pokatilov, L. I. Vereshchagin, O. N. Verkhozina, T. L. Petrova, A. G. Prodaikov, G. V. Ratovskii and O. V. Tyukalova, Russ. J. Appl. Chem., 2009, 82, 1769 CrossRef CAS.
- (a) S. G. Zlotin, A. I. Podgurskii, N. V. Airapetova and O. A. Lik'yanov, Russ. Chem. Bull., 1991, 40, 1460 CrossRef; (b) G. V. Malkov, A. V. Shastin, Y. I. Estrin, E. R. Badamshina and Y. M. Mikhailov, Propellants, Explos., Pyrotech., 2008, 33, 431 CrossRef CAS; (c) G. V. Malkov, A. V. Shastin, Y. I. Estrin, E. R. Badamshina and Y. M. Mikhailov, Russ. Chem. Bull., 2011, 60, 1940 CrossRef CAS; (d) S. Garg and J. M. Shreeve, J. Mater. Chem., 2011, 21, 4787 RSC; (e) P. Politzer, P. Lane and J. S. Murray, Cent. Eur. J. Energ. Mater., 2013, 10, 305 CAS; (f) A. V. Shastin and D. B. Lempert, Russ. J. Phys. Chem. B, 2014, 8, 716 CrossRef CAS.
- E. F. Witucki, G. L. Rowley, N. N. Ogimachi and M. B. Frankel, J. Chem. Eng. Data, 1971, 16, 373 CrossRef.
- (a) A. V. Shastin, T. I. Godovikova and B. L. Korsunskii, Russ. Chem. Rev., 2003, 72, 279 CrossRef CAS; (b) X. Zhang, Y. Li, W. Liu, Y. Yang, L. Peng and S. Pang, Chin. J. Energ. Mater., 2012, 20, 491 CAS; (c) V. V. Bakharev and A. A. Gidaspov, in Triazines: Synthesis, Applications and Toxicity, ed. R. B. Patel and A. Malhotra, Nova Science Publishers, New York, 2012, pp. 1–74 Search PubMed.
- (a) L. M. Kozlov and V. I. Burmistrov, Nitroalcohols and their Derivatives, Kazan, 1960 Search PubMed; (b) S. S. Novikov, M.-G. A. Shvekhgeimer, V. V. Sevostyanova and V. A. Shlyapochnikov, Chemistry of Aliphatic and Alicyclic Nitrocompounds, Khimiya, Moscow, 1974, pp. 56–114 Search PubMed.
- (a) M.-G. A. Shvekhgeimer, N. F. Pyatakov and S. S. Novikov, Usp. Khim., 1959, 28, 484 Search PubMed; (b) V. D. Nikolaev and M. A. Ishenko, Mendeleev Chem. J., 1997, 41(2), 15 Search PubMed; (c) M.-G. A. Shvekhgeimer, Russ. Chem. Rev., 1998, 67, 35 CrossRef.
- M. E. Sitzmann and W. H. Gilligan, J. Heterocycl. Chem., 1986, 23, 81 CrossRef CAS.
- D. F. Shellhamer, R. P. Callahan, V. L. Heasley, M. L. Druelinger and R. D. Chapman, Synthesis, 1997, 1056 CrossRef CAS.
- A. A. Gidaspov, V. V. Bakharev and I. K. Kukushkin, Russ. Chem. Bull., 2009, 58, 2154 CrossRef CAS.
- (a) A. A. Gidaspov, V. V. Bakharev, V. A. Zalomlenkov, P. S. Burkov, I. I. Artemyeva and I. A. Kuleshova, New Trends in Research of Energetic Materials, Proceedings of the Seminar, 16th, Pardubice, Czech Republic, 2013, p. 613 Search PubMed; (b) A. A. Gidaspov, V. V. Bakharev, V. A. Zalomlenkov, P. S. Burkov, M. N. Shaposhnikov and D. A. Bayzarov, New Trends in Research of Energetic Materials, Proceedings of the Seminar, 16th, Pardubice, Czech Republic, 2013, p. 617 Search PubMed.
- (a) D. Bartholomew, in Comprehensive Heterocycl. Chem. II, ed. A. R. Katritzky and C. W. Rees, Pergamon, Oxford, 1996, vol. 6, pp. 575–636 Search PubMed; (b) S. V. Chapyshev, E. N. Ushakov and A. V. Chernyak, Magn. Reson. Chem., 2013, 51, 562 CrossRef CAS PubMed; (c) V. A. Semenov, D. O. Samultsev and L. B. Krivdin, Magn. Reson. Chem., 2014, 52, 686 CrossRef CAS PubMed.
- (a) K. Yu. Suponitsky, K. A. Lyssenko, I. V. Ananyev, A. M. Kozeev and A. B. Sheremetev, Cryst. Growth Des., 2014, 14, 4439 CrossRef CAS; (b) A. B. Sheremetev, B. V. Lyalin, A. M. Kozeev, N. V. Palysaeva, M. I. Struchkova and K. Y. Suponitsky, RSC Adv., 2015, 5, 37617 RSC; (c) A. B. Sheremetev, I. L. Yudin, N. V. Palysaeva and K. Yu. Suponitsky, J. Heterocycl. Chem., 2012, 49, 394 CrossRef CAS.
- (a) C. B. Aakeroy, J. Desper, M. Fasulo, I. Hussain, B. Levin and N. Schultheiss, CrystEngComm, 2008, 10, 1816 RSC; (b) A. B. Sheremetev, N. V. Palysaeva, M. I. Struchkova, K. Yu. Suponitsky and M. Yu. Antipin, Eur. J. Org. Chem., 2012, 2266 CrossRef CAS; (c) A. Gavezzotti, CrystEngComm, 2013, 15, 4027 RSC.
- (a) A. B. Sheremetev, I. L. Yudin and K. Y. Suponitsky, Mendeleev Commun., 2006, 16, 264 CrossRef; (b) M. Gobel and T. M. Klapotke, Adv. Funct. Mater., 2009, 19, 347 CrossRef.
- A. I. Kitaigorodskii, Organic Chemical Crystallography, Consultants Bureau, New York, 1961 Search PubMed.
- M. Gobel and T. M. Klapotke, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63, o562 Search PubMed.
- M. Gobel and T. M. Klapotke, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2008, 64, o580 Search PubMed.
- A. Baumann, A. Erbacher, C. Evangelisti, T. M. Klapotke, B. Krumm, S. F. Rest, M. Reynders and V. Sproll, Chem.–Eur. J., 2013, 19, 15627 CrossRef CAS PubMed.
- Q. J. Axthammer, T. M. Klapotke, B. Krumm, R. Moll and S. F. Rest, Z. Anorg. Allg. Chem., 2014, 640, 76 CrossRef CAS.
- T. M. Klapotke, B. Krumm, S. F. Rest and M. Suceska, Z. Anorg. Allg. Chem., 2014, 640, 84 CrossRef.
- T. M. Klapotke, B. Krumm, R. Moll and S. F. Rest, Z. Anorg. Allg. Chem., 2011, 637, 2103 CrossRef.
- (a) F. H. Allen, Acta Crystallogr., 2002, B58, 380 CrossRef CAS; (b) Cambridge Structural Database, Version 5.36, 2014 Search PubMed.
- A. Kutoglu and E. Hellner, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 1617 CrossRef.
- Physics of explosion, ed. L. P. Orlenko, Fismatlit, Moscow, 3rd edn, 2002, vol. 1, p. 832 Search PubMed.
- J. B. Pedley, R. D. Naylor and S. P. Kirby, Thermodynamic Data of Organic Compounds, Chapman and Hall, 2nd edn, 1986 Search PubMed.
- D. Price, Chem. Rev., 1959, 59, 801 CrossRef CAS.
- V. I. Pepekin, M. N. Makhov and Yu A. Lebedev, Dokl. Akad. Nauk SSSR, 1977, 232, 852 CAS.
- G. B. Manelis, G. M. Nazin, Z. I. Rubtsov, and V. A. Strunin, Thermal Decomposition and Combustion of Explosives and Propellants, Taylor & Francis, NY, 2003 Search PubMed.
- (a) V. Y. Avdeyev, A. A. Gidaspov and Y. V. Moschenskiy, New Trends in Research of Energetic Materials, Proceedings of the Seminar, 15th, Pardubice, Czech Republic, 2012, p. 582 Search PubMed; (b) A. A. Gidaspov, E. V. Yurtaev, Y. V. Moschenskiy, V. Y. Avdeev and D. A. Karpov. New Trends in Research of Energetic Materials, Proceedings of the Seminar, 15th, Pardubice, Czech Republic, 2012, p. 613 Search PubMed.
- (a) F. Wang, H.-C. Du, J.-Y. Zhang and X.-D. Gong, J. Phys. Chem. A, 2011, 115, 11852 CrossRef CAS PubMed; (b) Y. Pan, W. Zhu and H. Xiao, J. Mol. Model., 2012, 18, 3125 CrossRef CAS PubMed; (c) X. Liang, Y. Zheng, B. Wang and X. Pu, Appl. Mech. Mater., 2013, 395–396, 104 CrossRef CAS.
- L. V. Kustova, E. P. Kirpichev, Y. I. Pubtsov, V. V. Avdonin, A. G. Korepin and L. T. Eremenko, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1981, 30, 1830 CrossRef.
- (a) S. W. Benson, F. R. Cruickshank, D. M. Golden, G. R. Haugen, H. E. O'Neal, A. S. Rodgers, R. Shaw and R. Walsh, Chem. Rev., 1969, 69, 279 CrossRef CAS; (b) Y. A. Lebedev, E. A. Miroshnichenko, and Y. K. Knobel, Thermochemistry of Nitro Compounds, Nauka, Moscow, 1970, p. 168 Search PubMed; (c) A. N. Kizin, P. L. Dvorkin, G. L. Ryzhova and Y. A. Lebedev, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1986, 35, 343 CrossRef; (d) E. V. Sagadeev, A. A. Gimadeev and V. P. Barabanov, Theor. Found. Chem. Eng., 2009, 43, 108 CrossRef CAS; (e) J. L. Holmes and C. Aubry, J. Phys. Chem. A, 2012, 116, 7196 CrossRef CAS PubMed.
- (a) S. P. Smirnov and A. S. Smirnov, Russ. J. Appl. Chem., 2009, 82, 1807 CrossRef CAS; (b) A. S. Smirnov, D. Lempert, T. S. Pivina and D. V. Khakimov, Cent. Eur. J. Energ. Mater., 2011, 8, 233 CAS.
- N. A. Kholevo, Sensitivity of explosives to impact, Mashinostroenie, Moscow, 1974, p. 232 Search PubMed.
- B. G. Trusov, in Proc. of the XIV Intern. Symp. on Chemical Thermodynamics, St-Petersburg, Russia, 2002, p. 483 Search PubMed.
- (a) J. P. Sutton and O. Biblarz, Rocket Propulsion Elements, 7th edn, Wiley, New York, 2001 Search PubMed; (b) N. Kubota, Propellants and Explosives, Wiley-VCH, Weinheim, 2002 Search PubMed; (c) A. Dey, A. K. Sikder, M. B. Talawar and S. Chottopadhyay, Cent. Eur. J. Energ. Mater., 2015, 12, 377 Search PubMed.
- M. B. Talawar, R. Sivabalan, T. Mukundan, H. Muthurajan, A. K. Sikder, B. R. Gandhe and A. Rao, J. Hazard. Mater., 2009, 161, 589 CrossRef CAS PubMed.
- D. B. Lempert, G. N. Nechiporenko and G. B. Manelis, Cent. Eur. J. Energ. Mater., 2006, 3(4), 73 Search PubMed.
- D. Kumari, R. Balakshe, S. Banerjee and H. Singh, Rev. J. Chem., 2012, 2, 240 CrossRef.
- (a) O. A. Luk'yanov and V. A. Tartakovskii, Ross. Khim. Zh., 1997, 41(2), 5 Search PubMed; (b) A. P. Vandel', A. A. Lobanova and V. S. Loginova, Russ. J. Appl. Chem., 2009, 82, 1763 CrossRef; (c) V. P. Sinditskii, V. Y. Egorshev, A. I. Levshenkov and V. V. Serushkin, J. Propul. Power, 2006, 22, 777 CrossRef CAS.
- D. B. Lempert, G. N. Nechiporenko and G. B. Manelis, Cent. Eur. J. Energ. Mater., 2011, 8, 25 CAS.
- G. Pavlovets and V. Tsutsuran, Physicochemical Properties of Powders and Propellants, Russian Ministry of Defense Publishing House, Moscow, 2009, p. 408 Search PubMed.
- (a) J. M. Berger and P. B. Butler, Combust. Sci. Technol., 1995, 104, 93 CrossRef CAS; (b) Y. D. Seo, S. H. Chung and J. J. Yoh, Fuel, 2011, 90, 1395 CrossRef CAS.
- E. A. Betterton, Crit. Rev. Environ. Sci. Technol., 2003, 33, 423 CrossRef CAS.
- (a) R. G. Schmitt, P. B. Butler and J. J. Freesmeier, Combust. Sci. Technol., 1997, 122, 305 CrossRef CAS; (b) S. R. Ganta, C. G. Miller, and G. K. Williams, Patent PCT WO 2008060366, 2008; (c) P. W. Leonard, D. E. Chavez, P. F. Pagoria and D. L. Parrish, Propellants, Explos., Pyrotech., 2011, 36, 233 CrossRef CAS; (d) D. L. Hordos, S. Domazet and S. P. Burns, US Pat., 8241444, 2012; (e) K. Hasue and K. Yoshitake, Sci. Technol. Energ. Mater., 2013, 74(3), 66 CAS.
- (a) A. B. Sheremetev, I. L. Yudin, N. N. Makhova, I. V. Ovchinnikov and D. B. Lempert, in Proc. 33st International Annual Conference of ICT – Energetic Materials: Synthesis, Production, and Application, Karlsruhe, FRG, 2002, p. 128/1–12 Search PubMed; (b) N. N. Makhova, A. B. Sheremetev, I. V. Ovchinnikov, I. L. Yudin, A. S. Ermakov, P. V. Bulatov, D. B. Vinogradov, D. B. Lempert and G. B. Manelis, in Proc. 35th International Annual Conference of ICT – Energetic Materials: Reactions of Propellants, Explosives and Pyrotechnics, Karlsruhe, FRG, 2004, p. 140/1–12 Search PubMed; (c) N. N. Makhova, A. S. Ermakov, I. V. Ovchinnikov, A. B. Sheremetev, I. L. Yudin, P. V. Bulatov, D. B. Vinogradov, V. A. Tartakovsky, D. B. Lempert, I. N. Zyuzin and G. B. Manelis, in Proc. 36th International Annual Conference of ICT &International Pyrotechnics Seminar-Energetic Materials: Performance and Safety, Karlsruhe, FRG, 2005, p. 185/1–10 Search PubMed.
- (a) C. Oommen and S. R. Jain, J. Hazard. Mater., 1999, 67, 253 CrossRef CAS PubMed; (b) V. P. Sinditskii, V. Yu. Egorshev, A. I. Levshenkov and V. V. Serushkin, Propellants, Explos., Pyrotech., 2005, 30, 269 CrossRef CAS; (c) V. P. Sinditskii, V. Y. Egorshev, D. Tomasi and L. T. DeLuca, J. Propul. Power, 2008, 24, 1068 CrossRef CAS.
- D. B. Lempert, G. B. Manelis, H. N. Nechiporenko and I. N. Zyuzin, in Proc. 7th International Symposium and Exhibition on Sophisticated Car Occupant Safety Systems, Karlsrue, FRG, 2004, p. 42 Search PubMed.
- O. A. Luk'yanov and G. V. Pokhvisneva, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1991, 40, 2439 CrossRef.
- V. A. Osyanin, D. V. Osipov, M. R. Demidov and Y. N. Klimochkin, J. Org. Chem., 2014, 79, 1192 CrossRef CAS PubMed.
- M. A. Epishina, I. V. Ovchinnikov, A. S. Kulikov, N. N. Makhova and V. A. Tartakovsky, Mendeleev Commun., 2011, 21, 21 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, copies of 1H, 13C, 14N, and 15N NMR spectra corresponding to all isolated compounds, and description of intermolecular interactions in the crystal. CCDC 1434738–1434740. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra05826d |
| This journal is © The Royal Society of Chemistry 2016 |