
Synthesis and characterization of calcium lanthanum sulfide via a wet chemistry route followed by thermal decomposition
Abstract
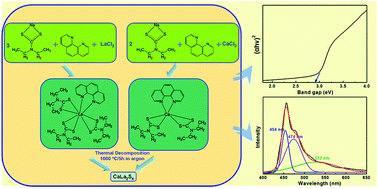
Calcium lanthanum sulfide (CaLa2S4) has been extensively studied as a promising candidate for advanced infrared optical ceramics. In the present research, we report the successful synthesis of CaLa2S4 via a wet chemistry method followed by thermal decomposition. CaLa2S4 precursor material was first prepared by a facile ethanol-based wet chemical single-source precursor route. The precursor was then thermally decomposed in argon at high temperature to form CaLa2S4. The phase composition and morphology of the synthesized CaLa2S4 powder were confirmed and observed by X-ray diffraction (XRD), scanning electron microscopy (SEM), and transmission electron microscopy (TEM), respectively. Surface area and pore size analyses showed that the CaLa2S4 powder had a high specific surface due to a combined effect of small particle size and the existence of mesopores. Optical characterization revealed that the synthesized CaLa2S4 powder exhibited quantum size confinement and near-band-edge photoluminescence.
 Please wait while we load your content...
Please wait while we load your content...

