
An efficient synthesis of 1-arylindazole-3-carboxamides using nitrile imines, isocyanides and 2-hydroxymethylbenzoic acid, followed by a chemoselective Buchwald–Hartwig intramolecular cyclization†
Abstract
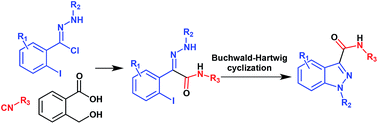
A convergent and efficient two-step synthesis of pharmaceutically relevant 1-arylindazole-3-carboxamides is reported. These molecules have been obtained in good to excellent yields (up to 98%) starting from a strategic reaction between isocyanides, 2-iodo-N-arylbenzohydrazonoyl chlorides and 2-hydroxymethylbenzoic acid followed by a chemoselective Buchwald–Hartwig intramolecular cyclization. This novel strategy provides an additional indazole synthesis to those already reported in literature both in the type of substrate as well as the substitution pattern obtainable in the products. Furthermore benzylisocyanide is herein reported for the first time as a convertible isocyanide providing an expeditious access to N-arylindazole-3-carbonitriles.
 Please wait while we load your content...
Please wait while we load your content...

