
Timing of calcium nitrate addition affects morphology, dispersity and composition of bioactive glass nanoparticles†
Abstract
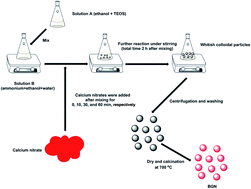
Bioactive glass nanoparticles (BGN) are promising materials for a number of biomedical applications. Many parameters related to the synthesis of BGN using sol–gel methods can affect their characteristics. In this study, the influence of timing of calcium nitrate (calcium precursor) addition during processing on BGN characteristics was investigated. The results showed that the addition timing could affect the morphology, dispersity and composition of BGN. With delayed addition of calcium nitrate, larger, more regular and better dispersed BGN could be synthesized while the gap between nominal and actual compositions of BGN was widened. However, the addition timing had no significant influence on structural characteristics, as BGN with different addition-timing of calcium nitrate exhibited similar infrared spectra and amorphous nature. The results also suggested that monodispersed BGN could be synthesized by carefully controlling the addition of calcium nitrate. The synthesized monodispersed BGN could release Si and Ca ions continuously for up to at least 14 days. They also showed in vitro bioactivity and non-cytotoxicity towards rat bone marrow-derived mesenchymal stem cells (rBMSCs). In conclusion, the timing of calcium precursor addition is an essential parameter to be considered when producing BGN which should exhibit monodisperse characteristics for biomedical applications.
 Please wait while we load your content...
Please wait while we load your content...

