
Dendrimer-functionalized LAPONITE® nanodisks loaded with gadolinium for T1-weighted MR imaging applications†
 *ac
*ac
Abstract
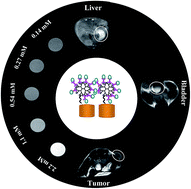
In this report, we present a facile approach to forming dendrimer-functionalized LAPONITE® (LAP) nanodisks loaded with gadolinium (Gd) for in vitro and in vivo T1-weighted MR imaging applications. In this work, LAP nanodisks were sequentially modified with silane coupling agents, succinic anhydride to have abundant carboxyl groups on their surface, and amine-terminated poly(amidoamine) (PAMAM) dendrimers of generation 2 (G2). The dendrimer-modified LAP nanodisks were then conjugated with gadolinium (Gd) chelator diethylenetriaminepentaacetic acid (DTPA), followed by Gd(III) chelation to form the LM–G2–DTPA (Gd) nanocomplexes. The designed LM–G2–DTPA(Gd) nanocomplexes were characterized via different techniques. Cell viability assay shows that the formed LM–G2–DTPA(Gd) nanodisks are non-cytotoxic in the given concentration range. With a high r1 relaxivity (2.05 mM−1 s−1), the LM–G2–DTPA(Gd) nanocomplexes are able to be used as an efficient contrast agent for T1-weighted MR imaging of cancer cells in vitro and animal organ/tumor model in vivo. The designed LM–G2–DTPA(Gd) nanocomplexes may hold a great promise to be used as a versatile nanoplatform for MR imaging of different biological systems.
 Please wait while we load your content...
Please wait while we load your content...

