
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric tandem hemiaminal-heterocyclization-aza-Mannich reaction of 2-formylbenzonitriles and amines using chiral phase transfer catalysis: an experimental and theoretical study†
Amedeo
Capobianco
*a,
Antonia
Di Mola
a,
Valentina
Intintoli
a,
Antonio
Massa
a,
Vito
Capaccio
a,
Lukas
Roiser
b,
Mario
Waser
b and
Laura
Palombi
*a
aDipartimento di Chimica e Biologia, Università di Salerno, Via Giovanni Paolo II, I-84084, Fisciano (SA), Italy. E-mail: acapobianco@unisa.it; lpalombi@unisa.it
bInstitute of Organic Chemistry, Johannes Kepler University Linz, Altenberger Strasse 69, 4040 Linz, Austria
First published on 24th March 2016
Abstract
The first asymmetric synthesis of 3-amino-substituted isoindolinones was accomplished via cascade hemiaminal-heterocyclization-intramolecular aza-Mannich reaction of benzylamines and 2-formylbenzonitriles using chiral phase transfer conditions (PTC). A theoretical study of the enantioselective step provides a rationale for the mode of action of the best performing phase transfer catalyst and the observed face selectivity.
Introduction
The 3-substituted-isoindolin-1-one framework belongs to a class of heterocyclic substructures which occurs in various natural and synthetic biologically active compounds.1 The isoindolinonic chiral core is present, for example, in the active part of commercially available pazinaclone, pagoclone and zopiclone, drugs that find applications as sedative-hypnotic agents.2 Isoindolinone nucleus is also found in natural and synthetic products with therapeutic applications in neurological disorders like Parkinson and Alzheimer diseases, as well as in the treatment of cancer, cardiac arrhythmia and HIV.3 Therefore, the interest in the construction of heterocycles based on the isoindolinonic ring is steadily increasing as testified by the large number of recent reports dealing with this subject.4In order to have small collections of potentially bioactive molecules focused on this heterocyclic architecture, in last years, we have also addressed research efforts in their achievement from readily available starting material 2-formylbenzonitriles using chemical5 or electrochemical6 strategies which enable tandem processes to directly access the molecular target.
In particular, we have recently demonstrated the possibility to induce the title reaction under electrochemical conditions, thus incorporating an interesting α-amino amidic functionality in the isoindolinonic ring.7
Furthermore, the product has been smoothly obtained in our laboratories, by simple treatment of the reactants with a stoichiometric amount of K2CO3 in CH3CN. In this case, as evidenced in Scheme 1 for a generic primary amine RNH2, the reaction should proceed by a sequence of hemiaminal formation (a), a base catalyzed heterocyclization step (b) and a subsequent rearrangement via an uncommon aza-Mannich reaction (c).
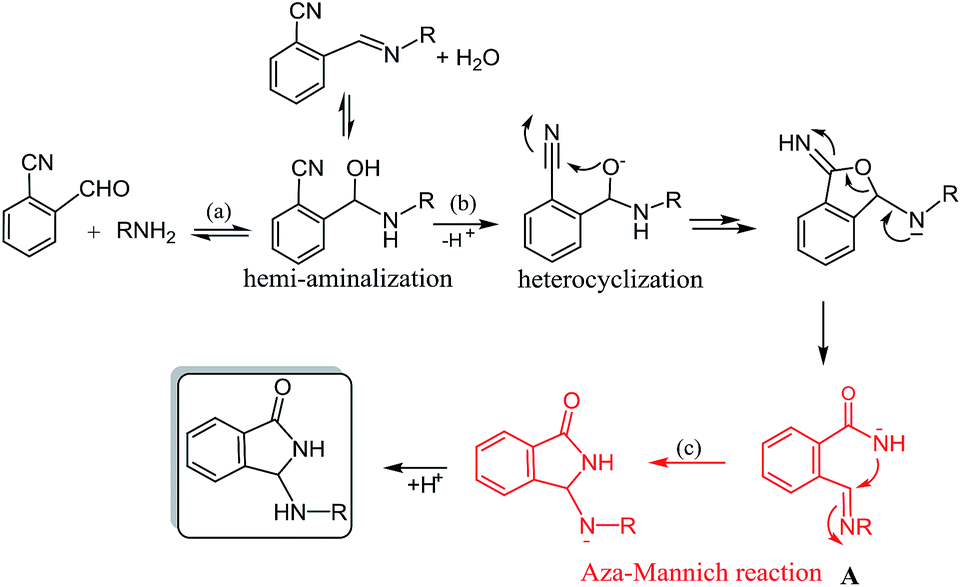 | ||
| Scheme 1 Reaction pathway for the reaction of 2-formybenzonitriles and primary amines. | ||
With the understanding of the basic principles of this chemistry in hand, we became interested to design an asymmetric version of this reaction, relying on the assumption that, under liquid–solid phase transfer conditions (i.e. organic solvent/inorganic base), chiral ammonium catalysts8 could help in the enantioselective construction of the chiral center during the rearrangement step.
In fact, the use of chiral-onium salts as phase transfer catalysts9 dates back a long time ago, nevertheless current research papers still provide new insights on this subject, both by reporting new potential uses in asymmetric synthesis10 and by offering explanations on their action mechanism.11
Herein we demonstrate the first asymmetric access to 3-amino-substituted isoindolinones via base catalyzed intramolecular aza-Mannich reaction, under chiral phase transfer conditions.
Results and discussion
In a first investigation (Table 1), to assess the effectiveness of the reaction, benzylamine and o-cyanobenzaldehyde were reacted in the presence of inorganic base K2CO3, using the series of ammonium salts listed in Fig. 1.12|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (% mol) | Time (h) | Yielda (%) | E.r.b |
| a Yields refer to isolated products. b E.r. determined by HPLC using a chiral stationary phase. c Reaction performed in toluene, 1a [50 mM]. d Reaction temperature: −20 °C. e Reaction performed in xylene, 1a [50 mM]. | ||||
| 1 | I (10) | 3 | 88 | 55/45 |
| 2 | II (10) | 3 | 88 | 56/46 |
| 3 | IV (10) | 3 | 96 | 57/43 |
| 4 | V (10) | 32 | 71 | 57/43 |
| 5 | QD-I (10) | 18 | 75 | 51/49 |
| 6 | QN-I (10) | 18 | 85 | 60/40 |
| 7c | VI (10) | 1 | 65 | 56/44 |
| 8c,d | VI (10) | 3 | 92 | 60/40 |
| 9c | III (10) | 18 | 92 | 40/60 |
| 10c | IV (10) | 1 | 86 | 65/35 |
| 11c | IV (5) | 1 | 54 | 72/28 |
| 12d,e | IV (10) | 15 | 81 | 64/36 |
| 13c,d | VII (10) | 8 | 92 | 32/68 |
| 14c,d | VIII (10) | 18 | 90 | 60/40 |
| 15c,d | IV (10) | 8 | 86 | 78/22 |
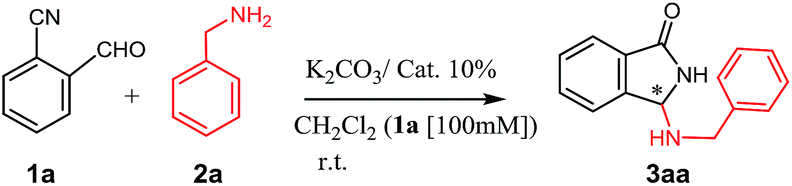
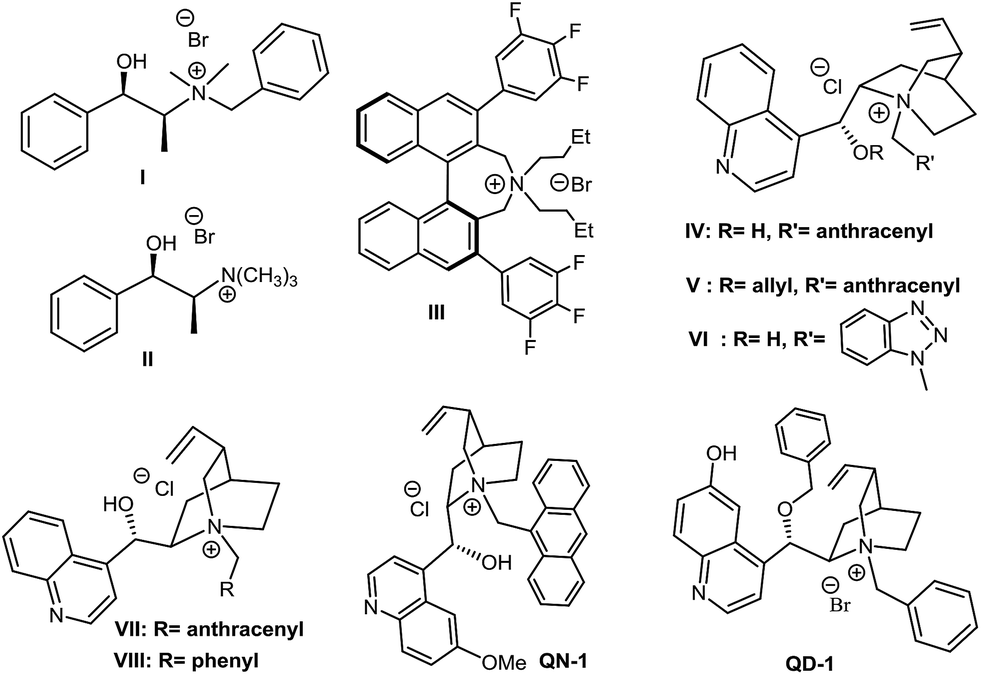 | ||
| Fig. 1 First screening of chiral ammonium salts tested in this work. | ||
Compared with classical monofunctional ammonium salts (III, V), bifunctional derivatives with a H-bonding donor (II, IV, VI, VII, VIII) show an improved catalytic activity (resulting in a higher conversion in shorter reaction times), while the presence of a bulky, rigidifying substituent on the nitrogen atom (i.e. 9-anthracenylmethyl group), appears to be crucial to achieve an increase in enantioselectivity. Despite many attempts have been made to optimize the reaction (choice of the solvent and base, different biphasic conditions, reactants and catalyst loading, change in temperature etc., see Chart 1 in the ESI†), the enantioselectivity was lying on modest levels with a maximum of 78/22 e.r. achieved under the conditions reported in Table 1, entry 15.
Although under phase transfer conditions, the stereocontrol usually derives from the electrostatic attraction of opposite charged species, several studies demonstrated that a further coordination via multiple noncovalent interactions (similarly to bifunctional organobase-H-bonding catalysts) may significantly enhance the catalyst performance in many cases.13 Based on the above observations, we decided to test the catalytic ability of ammonium salts having additional hydrogen-bonding sites (Fig. 2).14
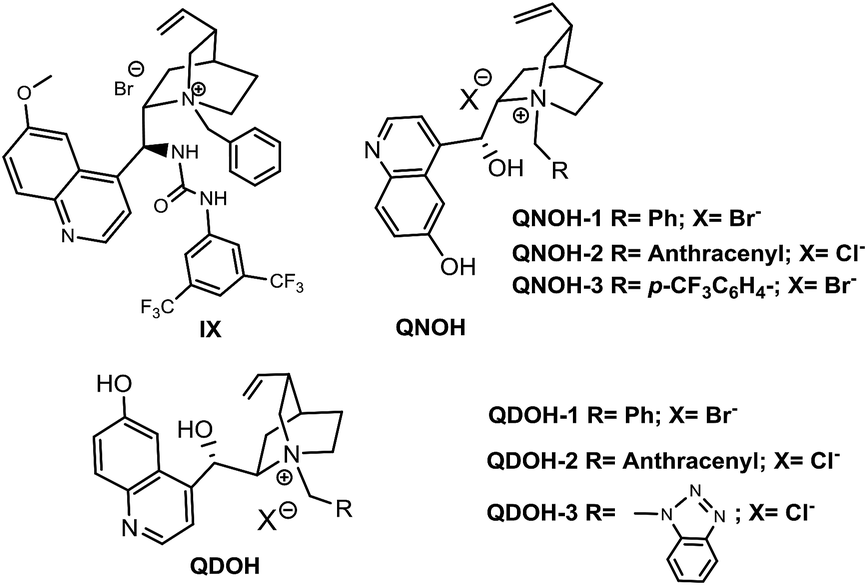 | ||
| Fig. 2 Multiple H-bonding donor containing chiral ammonium salts tested in this work. | ||
While Dixon's ammonium salt IX10d (Table 2, entry 1) performed the same way as catalysts of Table 1, placing the hydrogen-bond donor on the quinoline ring resulted in a substantial increase of enantioselectivity, QNOH-1 reaching the best enantiomeric ratio under optimized conditions (Table 2, entries 7 and 8).
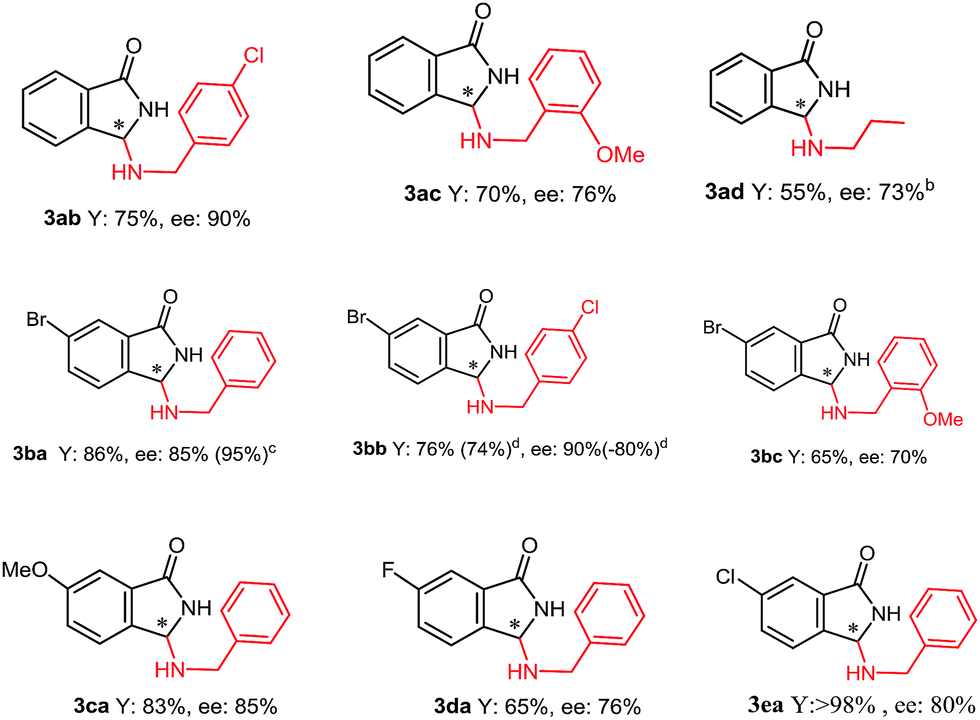
Furthermore, whereas ammonium salts QNOH-2 or QNOH-3 with different substituents on nitrogen atom gave comparable enantioselectivities, the crucial importance to have both the –OH functionalities emerges by comparison with the catalysts QN-1 and QD-1 which led to the expected product with low or vanishing enantiomeric excesses (Table 1, entries 6 and 7). Finally, it is worthy to note that the pseudo enantiomeric QDOH catalysts allowed an effective inversion of enantioselectivity which is not always observed with the first series of catalysts (Table 1, entry 14). Subsequently, the scope of the tandem process has been expanded to a variety of 2-formylbenzonitriles (1) and amines (2), using the most successful conditions obtained in Table 2, entry 7. As reported in Table 3, 2-formylbenzonitrile derivatives 1a–e with both electron-donating groups and electron-withdrawing groups smoothly reacted with primary amines 2a–d generating 3 in very good yields and ees (up to 90%). Noteworthy, an appreciable enantiomeric excess has been also obtained with aliphatic amine 2d.
| Entry | Catalyst (% mol) | Yieldsa (%) | E.r.b |
|---|---|---|---|
| a Yields refer to isolated products. b E.r. determined on HPLC equipped with chiral column. c Toluene was used as solvent, 8 h, r.t. d Nearly enantiopure 3aa was easily accessed through a reverse crystallization process from DCM/hexane solution.15 Indeed, as reported by us for other 3-substituted isoindolinones,5b the preferential formation of heterocrystals containing (R) and (S)-enantiomers, allowed the enantiomeric enrichment of mother liquor from the original non-racemic solution. | |||
| 1c | IX (10) | 92 | 39/61 |
| 2c | QDOH-1 (12) | 85 | 24/76 |
| 3c | QDOH-1 (6) | 85 | 24/76 |
| 4 | QDOH-1 (8) | 89 | 15/85 |
| 5 | QDOH-2 (8) | 84 | 12/88 |
| 6 | QDOH-3 (8) | 72 | 15/85 |
| 7c | QNOH-1 (8) | 92 | 87/13 |
| 8 | QNOH-1 (8) | 92 | 93/7 (99/1) d |
| 9 | QNOH-2 (10) | 92 | 92/8 |
| 10 | QNOH-3 (10) | 92 | 92/8 |
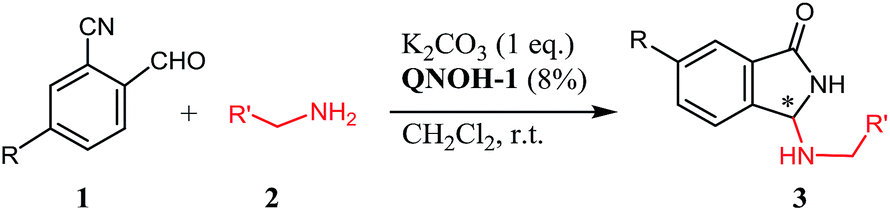
Mechanism
Given these interesting results, to unravel the origin and the sense of the asymmetric induction, we have investigated the enantioselective step of the reaction of 1a with 2a (i.e. the aza-Mannich reaction) by quantum chemical computations.Our study started by analyzing the conformer distribution of the cinchona alkaloid-derivative ammonium cation QNOH-1 and the A intermediate involved in the rearrangement step. In line with previous results,16 the ammonium cation is predicted to assume open conformations at 298 K with a preference for the anti orientation (see Fig. S1 and Table S1 in the ESI†), the closed forms being ruled out by unfavorable steric interactions between the benzyl and quinoline moieties.
All the bonds of the A intermediate were allowed to be rotatable in docking simulations. In this way also the possibility of the Z configuration was considered for the binary complexes formed by the A intermediate and the ammonium catalyst. Sampling the full conformational space of such a highly flexible system was a necessary effort to make sure that the most significant arrangements for the A:QNOH-1 ion-pairs were actually located (see Table S2 in the ESI†).17
Because of the large size of the system, a plethora of binary complexes between the QNOH-1 ammonium and the A intermediate has to be expected and indeed 74 unique complexes were found by DFT computations (Tables S3 and S4 of the ESI†). Moreover the presence of two hydroxyl donor groups on the catalyst and three acceptor units on the A intermediate gives rise to a wide panel of association modes. The most populated binary complexes are characterized by two hydrogen bonds, according to the coordination patterns exemplified by Ia1 (and Ia2), Ib1 (and Ib2), Ic and Id reactant complexes of Fig. 3 (a, b, c and d denote the hydrogen bonds patterns; additional arrangements are reported in Fig. S2 of the ESI†). In Ia1 and its nearly isoenergetic homologue Ia2 (see Table S5 in the ESI†) the nucleophilic nitrogen of intermediate A is engaged in a H bond with the hydroxyl at the C9 of QNOH-1 while the iminic nitrogen holds the hydroxyl at the C6′. An opposite coordination is found for Ic; here the C9 hydroxyl holds the iminic nitrogen while the C6′ hydroxyl coordinates the nucleophilic nitrogen of A. Both the E configuration of A and the open conformation of the catalyst are retained in Ia1, Ia2 and Ic (Fig. 4).
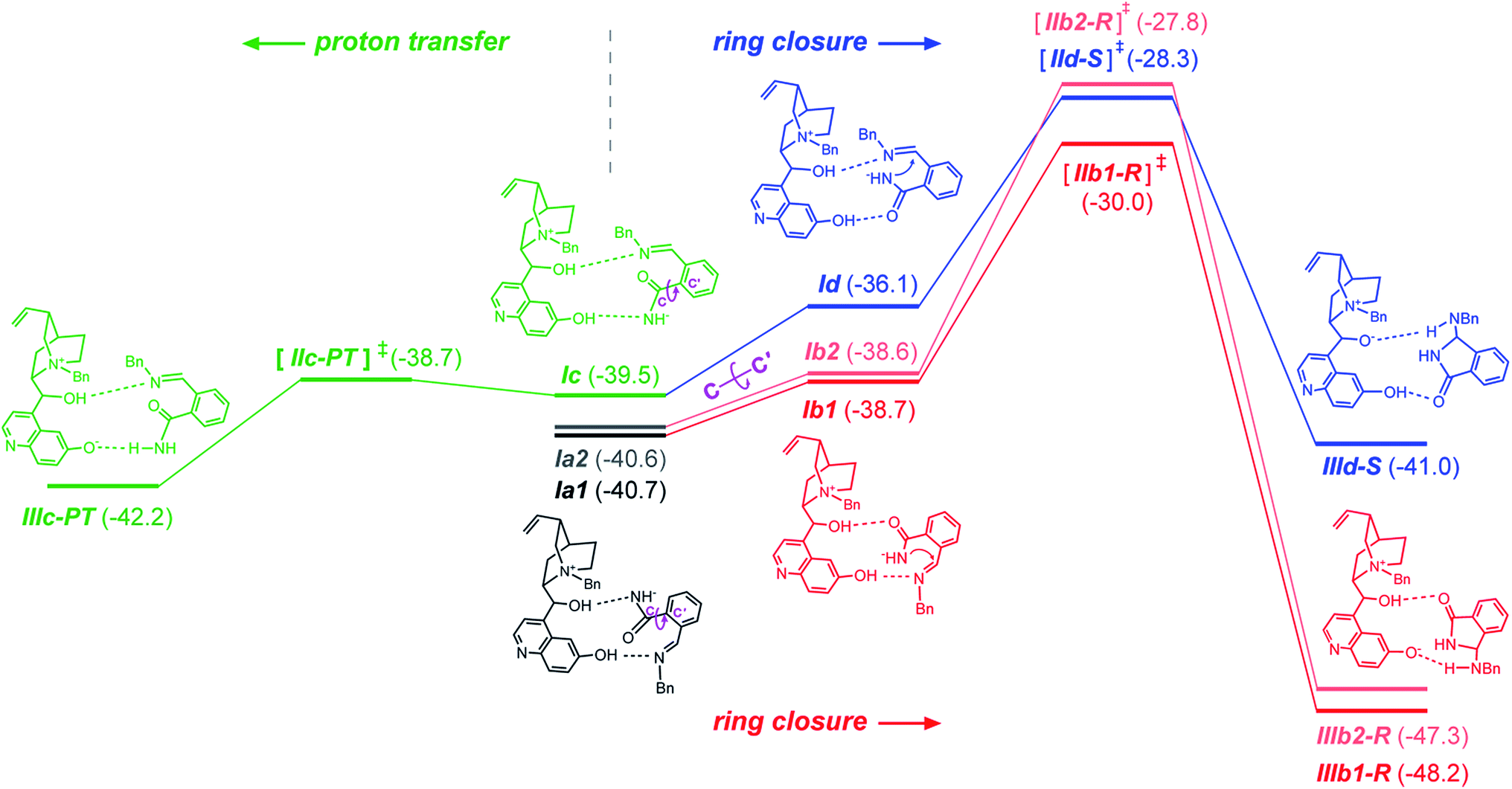 | ||
| Fig. 3 Schematic energy profile of the enantioselective ring closure step (red and blue) and proton transfer (green). Relative energy (kcal mol−1) refers to non-interacting A intermediate and QNOH-1 ammonium cation. | ||
 | ||
| Fig. 4 The most stable binary complexes of the A intermediate and QNOH-1 cation for a and c patterns. Noncritical hydrogen atoms omitted for clarity. Distances in Å. | ||
They are strongly associated ion-pairs further stabilized by hydrogen bonds and π–π stacking interactions between the aromatic moiety of the intermediate and the quinoline ring of the catalyst. Ia1 is slightly favored over Ic due to a T-shaped CH–π interaction involving the phenyl rings of A and QNOH-1 (Fig. 4).
Starting from Ia1, Ia2, and Ic, a torsion of 180° about the C–C′ bond (Fig. 3) generates Ib1, Ib2 and Id, respectively, in which the carbonyl oxygen replaces the nucleophilic nitrogen as the acceptor. Ib1, Ib2 and Id rather than Ia1, Ia2 and Ic are the actual reactive complexes for the ring closure process, because in Ib1, Ib2 and Id the nucleophilic nitrogen comes into close contact with the electrophilic carbon (Fig. 3). Indeed, following the reaction coordinate for the ring-closure, Ib1 and Ic evolve into IIb1-R and IId-S transition states in which an incipient formation of the C–N bond occurs, as revealed by the shortening of the C–N distance, which amounts to ca. 3 Å in Ib1 and 1.9 Å in IIb1-R (Fig. 5).
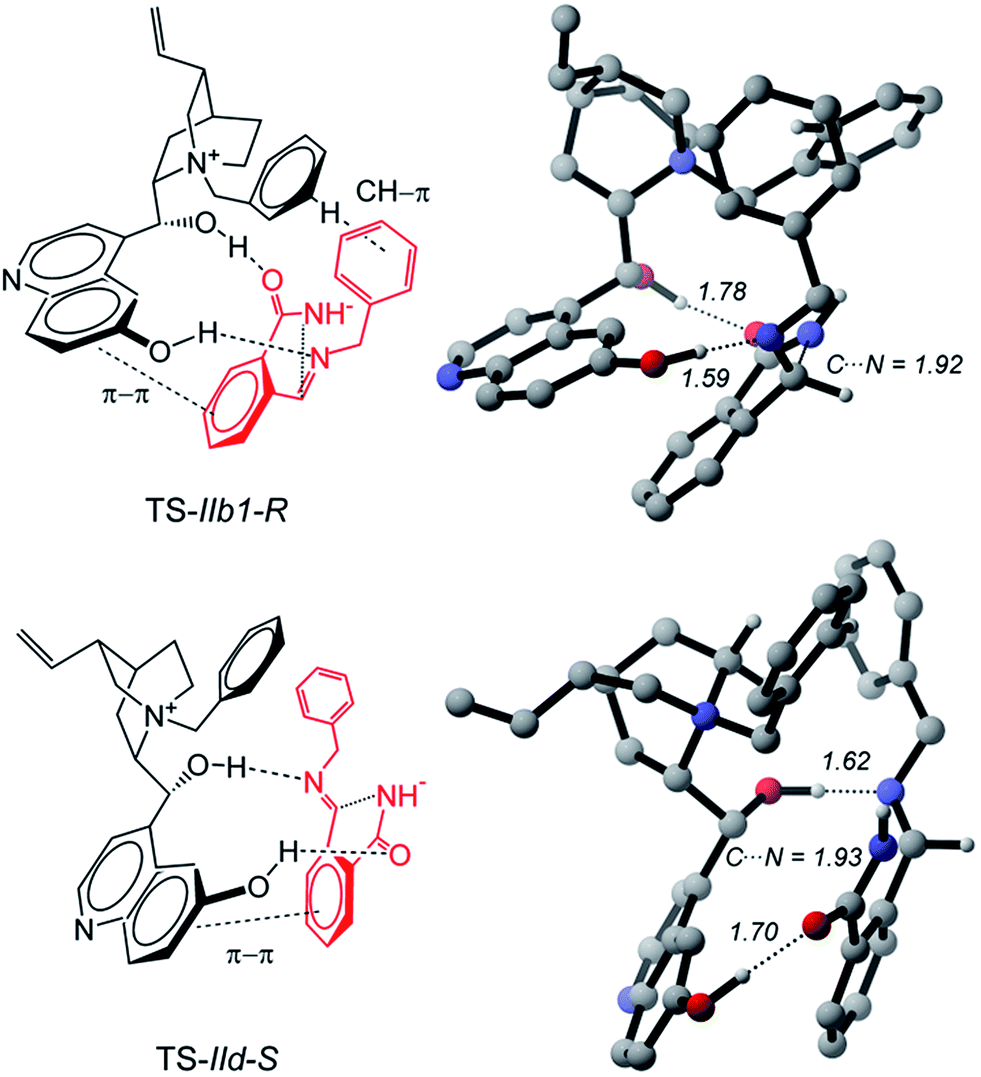 | ||
| Fig. 5 Energetically most accessible transition states leading to the R and S enantiomers of product 3aa. Noncritical hydrogen atoms omitted for clarity. Distances in Å. | ||
IIb1-R and IId-S are predicted to be the most stable among all pro-R and pro-S transition states, (see Table S3 in the ESI†). IIb1-R is more stable than IId-S by 1.7 kcal mol−1 (Fig. 3) implying an easier accessibility for the R configuration of the product. This is confirmed by a statistical analysis considering all the transition states (see Table S3 of ESI†) which gives an 86% ee in favor of the R enantiomer, in very good agreement with the observed excess. The nucleophilic attack is accompanied by an increase of negative charge on the imine nitrogen (step b of Scheme 1). This is confirmed by computations: the APT charge of the imine nitrogen charge passes from −0.9 in Ib1 and Id to −1.4 in IIb1-R and IId-S thus showing that the proton involved in the O–H⋯N hydrogen bond greatly facilitates the ring closure step.
Therefore, the different stability of the transition states can be traced back to the different patterns of hydrogen bonds. In fact it becomes relevant whether the quinoline OH or the C9 hydroxyl of the catalyst acts as the donor in the OH⋯N bond because the Lewis basicity of the imine moiety of A in the transition state is supported by a quite strong phenol-like acidic function in the former case (IIb1-R), while a much weaker acidic hydroxyl is involved in the latter case (IId-S). Following the intrinsic reaction coordinate, IIb1-R and IId-S evolve into IIIb1-R and IIId-S product complexes, where the C–N bond is fully established. Very interestingly, the ring closure is predicted to proceed in concert with a proton transfer from the ammonium catalyst to the imine nitrogen, in such way that the final product 3 is directly obtained and the ammonium catalyst is converted into its corresponding betaine (Fig. 6). This strongly suggests that the initial base activator is indeed the betaine species, whose occurrence can be reasonably expected due to the acidity of QNOH-1 ammonium and the presence of carbonate.18
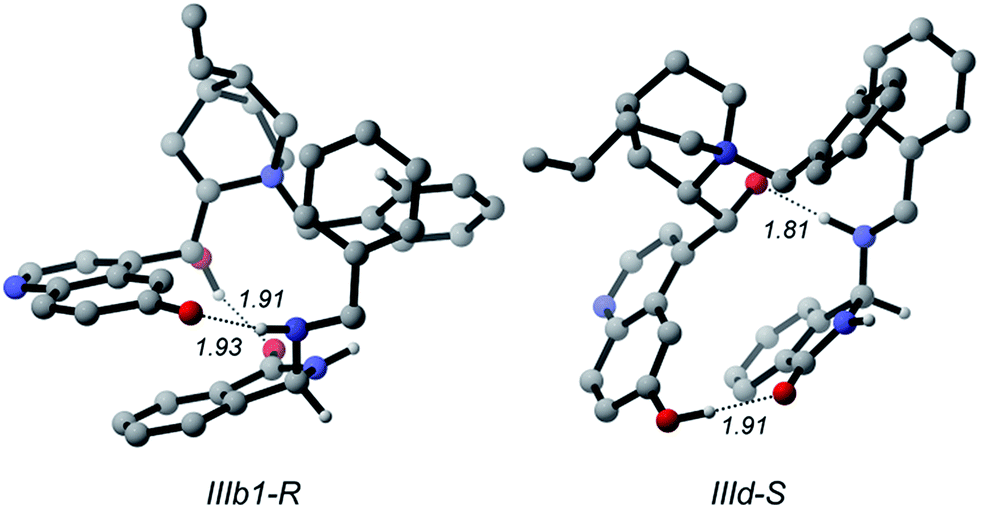 | ||
| Fig. 6 R and S enantiomers of the final product coordinated to betaine. Noncritical hydrogen atoms omitted for clarity. Distances in Å. | ||
The final step is thermodynamically favored for the pathway leading to R product, IIIb1-R being 7.5 kcal mol−1 more stable than Ia (Fig. 3). At variance with IIIb1-R where the proton transfer involves the acidic phenol hydroxyl of quinoline, in IIId-S deprotonation of QNOH-1 ammonium occurs on C9 hydroxyl, making the ring formation for the S-configured product a less affordable process.
We have also considered the possibility of a direct acid–base reaction between the rather acidic QNOH-1 cation and intermediate A. That product would lead to the amide depicted in Fig. 3 (bottom left, green bars). First of all (see Fig. S2 in the ESI†), proton transfer is predicted to be a viable process only for Ic (Fig. 3) because only there the most acidic function of the catalyst can interact with the most basic function of the A intermediate. From Ic a proton transfer from QNOH-1 to A could result into IIIc-PT product complex in which amide is coordinated to the betaine. However a glance at Fig. 3 (green bars, bottom left) will show that formation of the amide, although kinetically feasible, would lead to a very modest energy gain (1.5 kcal mol−1) in comparison with the ring closure process (7.5 kcal mol−1). In addition, the occurrence of two affordable and nearly isoenergetic pathways (dark and light red, Fig. 3) leading to the ring closure for the R product further favours the formation of product III.
In summary the theoretical investigation confirms the expectations of the design process, stressing the fundamental role of the hydroxyls on selectivity: the presence of two OH groups makes the catalyst a bifunctional promoter capable of orienting the intermediate A in a preferential way through the establishment of an effective network of hydrogen bonds, which happens to facilitate the R-configured product.
On the bases of available evidence18 and DFT results we can assume that, after the initial hemi-aminalization, the catalytic cycle should start with an acid–base reaction between the ammonium cation and CO32− with the generation of the ammonium betaine assisting the heterocyclization and/or the rearrangement step by H-bonding catalysis. The rearrangement step produces the structured intermediate A/ammonium cation ion-pair that undergoes the asymmetric aza-Mannich reaction. The asymmetric step leads to the final product 3 regenerating the betaine catalyst (Scheme 2).
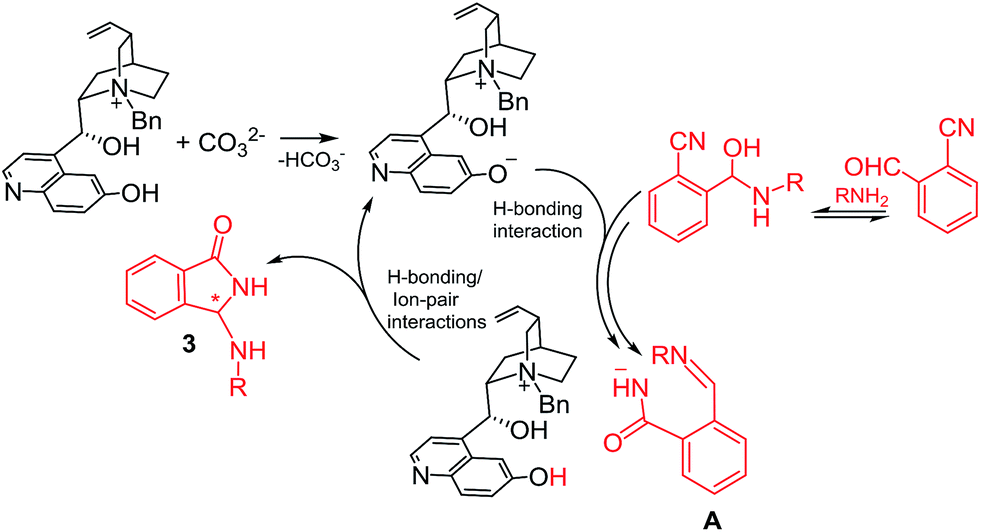 | ||
| Scheme 2 Comprehensive catalytic cycle. | ||
Assignment of the absolute configuration of the product
Since various attempts to crystallize product 3aa failed, in order to assess the absolute configuration of the major enantiomer, the chiral amines (S)-2f and (R)-2f were reacted with 1a in the presence of QNOH-1 as catalyst (Fig. 7).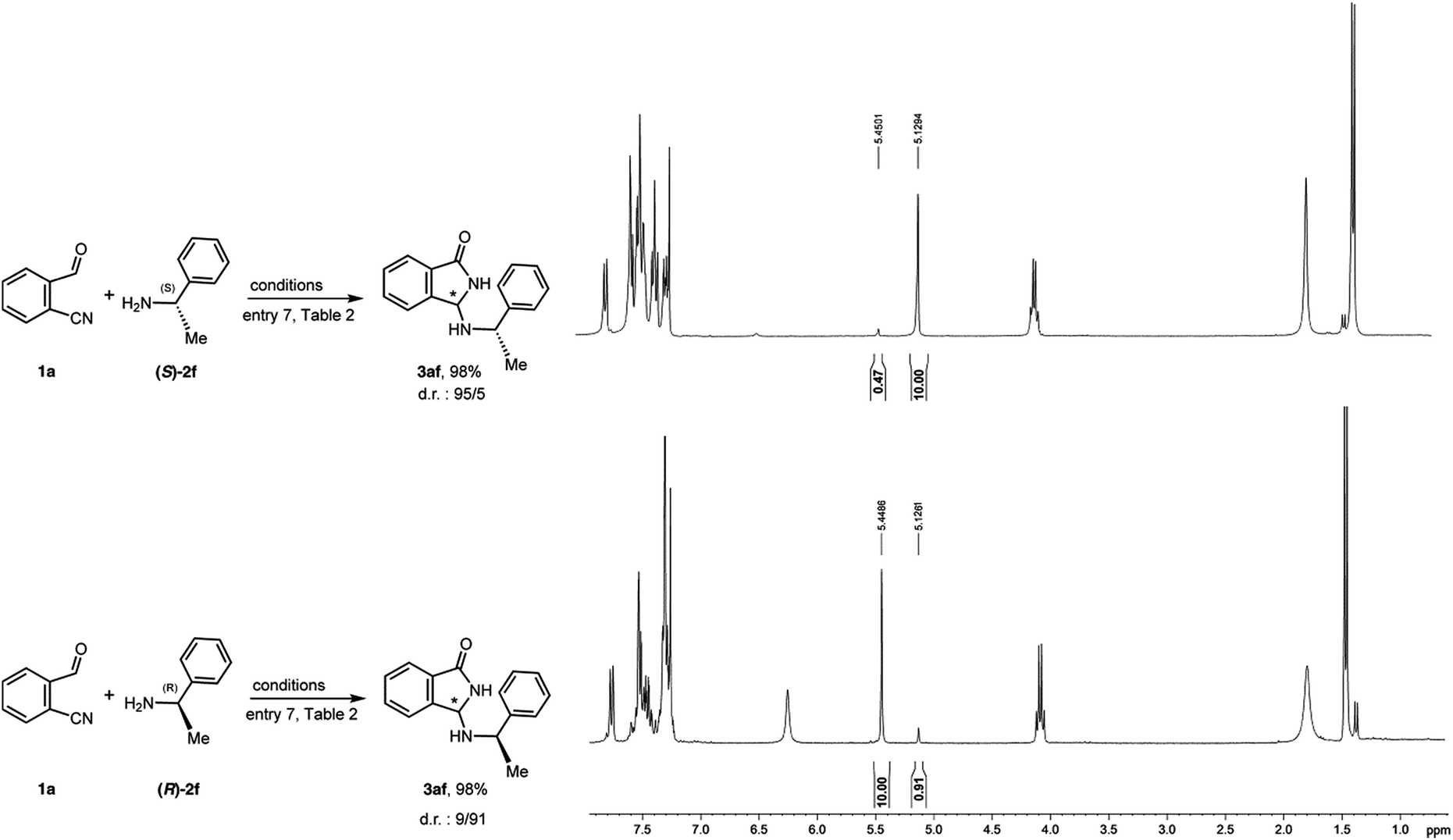 | ||
| Fig. 7 Reaction of 1a with chiral amines (S)-2f and (R)-2f under phase transfer conditions and relative 1H-NMR spectra of the diastereomeric mixtures. | ||
As evidenced by the NMR spectra on the crude mixtures, (S)-2f and (R)-2f yielded the product 3af with reverse diastereoselectivity, thus implying the same configuration at the newly created stereogenic center. Moreover, in both cases, the diastereoselectivity is comparable to the enantioselectivity obtained with achiral benzylamine 2a. On the contrary, a nearly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereoisomers is obtained under non-asymmetric reaction conditions. These results let us deduce that, also with chiral amines, the stereocontrol is totally due to the catalyst and, as a consequence, the absolute configuration of 3aa can be confidently attributed on the base of the configurations of 3af. The relative configurations of the two diastereoisomers 3af were deduced from the 1H NMR spectra which were assigned with the aid of computations. The chemical shift of the H atom on the isoindolinonic ring is predicted to be 5.42 ppm for the diastereoisomer (RR,SS) and 5.05 ppm for the diastereomer (SR,RS).
:1 mixture of diastereoisomers is obtained under non-asymmetric reaction conditions. These results let us deduce that, also with chiral amines, the stereocontrol is totally due to the catalyst and, as a consequence, the absolute configuration of 3aa can be confidently attributed on the base of the configurations of 3af. The relative configurations of the two diastereoisomers 3af were deduced from the 1H NMR spectra which were assigned with the aid of computations. The chemical shift of the H atom on the isoindolinonic ring is predicted to be 5.42 ppm for the diastereoisomer (RR,SS) and 5.05 ppm for the diastereomer (SR,RS).
Taking advantage from the known configuration of the chiral starting amines, the comparison with the experimental spectra (δ = 5.45 ppm and 5.13 ppm) unequivocally shows that the new formed stereocenter possesses R configuration.
This strongly suggests that the most abundant enantiomer of compound 3aa also possesses R configuration, as anticipated by DFT predictions.
Experimental
For detailed experimental information, spectra and chromatograms of all the new compounds see the ESI.† Catalysts I, II, III, IV, V and VII are commercially available. Catalysts VI,19QN1,13aQD1 (ref. 20) and IX10d have been prepared in accordance with the literature and gave spectral and analytical data as reported.General procedure for the synthesis of bifunctional chiral phase transfer catalysts QDOH-1, QDOH-2, QNOH-1, QNOH-2, QNOH-3
All the ammonium salts were prepared by following the reported procedure for the quaternisation of tertiary amines13a from 6′-demethylated quinine (catalyst QNOH) and 6′-demethylated quinidine (catalyst QDOH) which were prepared as reported in the literature.21General procedure for the synthesis of isoindolinones 3 under phase transfer catalyzed conditions
In a round-bottom flask, amine 2 (0.11 eq., 0.11 mmol) was added at room temperature to a stirred solution of 2-formylbenzonitrile 1 (0.10 mmol), K2CO3 (1 eq., 0.1 mmol) and phase transfer catalyst QNOH-1 (8% mol) in CH2Cl2 (1.8 mL). The mixture was stirred until starting material disappeared (8–18 h), then the solvent was removed under reduced pressure and the residue purified by flash chromatography on silica gel with hexane–ethyl acetate 1:1 mixtures.
:1, c = 0.5, CHCl3, 22 °C). Spectra and analytical data as reported.7 HPLC: Chiral pack AD column, hexane–iPrOH 8:2, 0.6 mL min−1, λ = 254 nm (tminor = 12.5 min, tmajor = 15.7 min).
:5, c = 0.5, CHCl3, 17 °C). Spectra and analytical data as reported.7 HPLC: Chiralpack AD column, hexane–iPrOH 8:2, 0.6 mL min−1, λ = 254 nm (tminor = 15.5 min, tmajor = 16.6 min).
:2, 0.6 mL min−1, λ = 254 nm (tminor = 13.0 min, tmajor = 15.7 min).
:2, 0.6 mL min−1, λ = 254 nm (tminor = 9.0 min, tmajor = 11.3 min).
:2, c = 0.4, CHCl3, 19 °C). Spectra and analytical data as reported.7 HPLC: Chiralpack AD column, hexane–iPrOH 8:2, 0.6 mL min−1, λ = 254 nm (tminor = 13.0 min, tmajor = 15.7 min).
:7, c = 0.8, CHCl3, 19 °C). Pale yellow amorphous solid; 1H NMR (300 MHz, CDCl3): 7.94 (1H, s); 7.72–7.68 (1H, m); 7.52–7.46 (2H, m); 7.28–7.26 (3H, m); 5.49 (1H, s); 4.03 (1H, bs); 3.85 (1H, d, J = 13.3 Hz); 3.71 (1H, d, J = 13.3 Hz); 2.28 (1H, bs). 13C NMR (75 MHz, CDCl3): 143.9; 137.7; 135.3; 134.1; 133.1; 129.4; 128.7; 126.7; 125.4; 123.5; 69.8; 47.8. MS (ESI): m/z = 273 (M + H+). Anal. calcd for C15H13ClN2O: C, 66.06; H, 4.80; Cl, 13.00; N, 10.27; O, 5.87. HPLC: Chiralpack AD column, hexane–iPrOH 8:2, 0.6 mL min−1, λ = 254 nm (tmajor = 18.3 min, tminor = 23.4 min).
:2, 0.6 mL min−1, λ = 254 nm (tminor = 12.7 min, tmajor = 17.0 min).
:2, 0.6 mL min−1, λ = 254 nm (tminor = 14.4 min, tmajor = 19.7 min).
:2, 0.6 mL min−1, λ = 254 nm (tminor = 11.0 min, tmajor = 12.7 min).
:10, c = 0.5, CHCl3, 17 °C). White amorphous solid; 1H NMR (400 MHz, CDCl3): 7.79 (1H, s); 7.56 (2H, bs); 7.34–7.28 (5H, m); 5.53 (1H, s); 3.90 (1H, d, J = 13.0 Hz); 3.75 (1H, d, J = 13.0 Hz); 1.98 (1H, bs). 13C NMR (100 MHz, CDCl3): 168.9; 143.6; 139.3; 135.6; 133.9; 132.5; 128.6 (2C); 128.1 (2C); 127.5; 125.1; 123.7; 69.8; 48.8 MS (ESI): m/z = 295 (M + Na+). Anal. calcd for C15H13ClN2O: C, 66.06; H, 4.80; Cl, 13.00; N, 10.27; O, 5.87. Found: C, 66.09; H, 4.82; Cl, 12.96; N, 10.25. HPLC: Chiralpack AD column, hexane–iPrOH 8:2, 0.6 mL min−1, λ = 254 nm (tminor = 13.9 min, tmajor = 16.4 min).
Computational procedure
Conformer search for the QNOH-1 ammonium cation, intermediate A and 3af (RR and RS diastereoisomers) was carried out by using the MMFF94 force field as implemented in Spartan.22 Different starting structures were considered for each system and both the systematic and the Montecarlo based algorithms were employed. All the conformers up to 10 kcal mol−1 with respect to minimum energy structures were then optimized by quantum chemical computations using density functional theory (DFT). Guess structures of the ion pair consisting of catalyst QNOH-1 and intermediate A were obtained by docking simulations employing a homemade program; the starting geometries of the catalyst and the ligand were initially optimized at the MMFF94 level. Geometry optimizations and Hessian computations needed to ascertain the nature of the stationary points were carried out by using the M06-2X functional which is known to provide reliable geometries and barrier heights especially for large systems.23 The 6-31G(d) basis set was used in optimizations; single point computations with the 6-311+G(d,p) basis set were carried out to obtain accurate energies. Intrinsic reaction coordinate (IRC) calculations were performed to check the connectivity of all the TS with their corresponding minima. All the TS's exhibit only one large imaginary frequency corresponding to the stretching coordinate of the forming bond, while the minimum energy structures have only positive eigenvalues of the Hessian matrix. Solvent effects were taken into account by means of the polarizable continuum model (PCM) which was included in all computations.24 All the quoted energies refer to 6-311+G(d,p) basis set; they include zero point vibrational corrections (computed at the 6-31G(d) level) and the solvent polarization contribution. Atomic charges were estimated by the atomic polar tensor approach (APT).25 DFT computations were carried out by using the Gaussian program.26Although entropic contributions are expected to be significant for association processes, their effects should elide in differences, the quantities that really matter. Therefore our discussion has been based on internal energies, following the same arguments given in ref. 27.
NMR computations for the RS/RR diastereomers of 3af were carried out by following the procedure of ref. 28: the minimum energy configurations and the energetics needed for the Boltzmann weighting of the shielding tensors were taken from PCM(CHCl3)/M06-2X/6-31+G(d,p) calculations, while shielding tensors were calculated at the PCM(CHCl3)/B3LYP/6-311+G(2d,p) level.
Conclusions
The first asymmetric route to chiral N-Mannich bases of 1-isoindolinones has been successfully opened by using a readily available bifunctional cinchona-derivative ammonium salt as phase transfer catalyst in the presence of K2CO3 as base. The goal was achieved thanks to an efficient cascade reaction of 2-formylbenzonitriles and primary amines involving an uncommon enantioselective aza-Mannich rearrangement step. Theoretical computations show that QNOH-1 acts as bifunctional catalyst determining the enantio-sense of the aza-Mannich step through the establishment of an effective network of hydrogen bonds.Acknowledgements
This research was funded by Region Campania under POR Campania FESR 2007–2013 – O.O. 2.1 (FarmaBioNet). The financial support of the University of Salerno is gratefully acknowledged.Notes and references
- (a) H. Kawagishi, M. Ando and T. Mizuno, Tetrahedron Lett., 1990, 31, 373 CrossRef CAS; (b) S. F. Hinkley, J. C. Fettinger, K. Dudley and B. B. Jarvis, J. Antibiot., 1999, 52, 988 CrossRef CAS PubMed; (c) M. Jacolot, M. Jean, N. Tumma, A. Bondon, S. Chandrasekhar and P. Van de Weghe, J. Org. Chem., 2013, 78, 7169 CrossRef CAS PubMed; (d) E. Li, L. Jiang, L. Guo, H. Zhang and Y. Che, Bioorg. Med. Chem., 2008, 16, 7894 CrossRef CAS PubMed; (e) N. Slavov, J. Cvengroš, J. M. Neudörfl and H.-G. Schmalz, Angew. Chem., Int. Ed., 2010, 49, 7588 CrossRef CAS PubMed.
- (a) I. Takahashi, T. Kawakami, E. Hirano, H. Yokota and H. Kitajima, Synlett, 1996, 353 CrossRef CAS; (b) t. L. Stuk, B. K. Assink, R. C. Bates Jr, T. R. Erdman, V. Fedij, S. M. Jennings, J. L. Lassig, R. J. Smith and L. T. Smith, Org. Process Res. Dev., 2003, 7, 851 CrossRef CAS; (c) E. Van der Kleijn, Eur. J. Clin. Pharmacol., 1989, 36, 247 CrossRef CAS PubMed.
- (a) H. A. Priestap, Phytochemistry, 1985, 24, 848 Search PubMed; (b) A. Couture, E. Deniau, P. Grandclaudon and C. Hoarau, J. Org. Chem., 1998, 63, 3128 CrossRef CAS; (c) A. Mertens, J. H. Zilch, B. Konig, W. Schafer, T. Poll, W. Kampe, H. Seidel, U. Leser and H. Leinert, J. Med. Chem., 1993, 36, 2526 CrossRef CAS PubMed; (d) A. Bjoere, et al. WO2008008022, 2008.
- For recent reviews on the synthesis of isoindolinones, see: (a) A. Di Mola, L. Palombi and A. Massa, Targets Heterocycl. Syst., 2014, 18, 113 CAS; (b) K. Speck and T. Magauer, Beilstein J. Org. Chem., 2013, 9, 2048 CrossRef PubMed.
- (a) A. Di Mola, M. Tiffner, F. Scorzelli, L. Palombi, R. Filosa, P. De Caprariis, M. Waser and A. Massa, Beilstein J. Org. Chem., 2015, 11, 2591 CrossRef CAS PubMed; (b) M. Perillo, A. Di Mola, R. Filosa, L. Palombi and A. Massa, RSC Adv., 2014, 4, 4239 RSC; (c) C. Petronzi, S. Collarile, G. Croce, R. Filosa, P. De Caprariis, A. Peduto, L. Palombi, V. Intintoli, A. Di Mola and A. Massa, Eur. J. Org. Chem., 2012, 5357 CrossRef CAS; (d) V. More, R. Rohlmann, O. G. Mancheno, C. Petronzi, L. Palombi, A. De Rosa, A. Di Mola and A. Massa, RSC Adv., 2012, 2, 3592 RSC.
- (a) L. Palombi, A. Di Mola, C. Vignes and A. Massa, Mol. Diversity, 2014, 18, 323 CrossRef CAS PubMed; (b) P. Antico, V. Capaccio, A. Di Mola, A. Massa and L. Palombi, Adv. Synth. Catal., 2012, 354, 1717 CrossRef CAS.
- L. Palombi, A. Di Mola and A. Massa, New J. Chem., 2015, 39, 81 RSC.
- (a) M. J. O'Donnell, W. D. Bennett and S. Wu, J. Am. Chem. Soc., 1989, 111, 2353 CrossRef; (b) E. J. Corey, F. Xu and M. C. Noe, J. Am. Chem. Soc., 1997, 119, 12414 CrossRef CAS; (c) R. Helder, J. C. Hummelen, R. W. P. M. Laane, J. S. Wiering and H. Wynberg, Tetrahedron Lett., 1976, 17, 1831–1834 CrossRef; (d) U. H. Dolling, P. Davis and E. J. J. Grabowski, J. Am. Chem. Soc., 1984, 106, 446–447 CrossRef CAS.
- For recent reviews on chiral phase transfer catalysts see: (a) S. Kaneko, Y. Kumatabara and S. Shirakawa, Org. Biomol. Chem., 2016 10.1039/c5ob02446c; (b) S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312 CrossRef CAS PubMed; (c) S.-s. Jew and H.-g. Park, Chem. Commun., 2009, 7090 RSC; (d) Asymmetric Phase Transfer Catalysis, K. Maruoka, ed. Wiley-VCH, Weinheim, 2008 Search PubMed; (e) K. Maruoka, Chem. Rec., 2010, 10, 254 CrossRef CAS PubMed.
- For selected recent examples, see: (a) R. Herchl and M. Waser, Tetrahedron, 2014, 70, 1935–1960 CrossRef CAS; (b) J. Novacek and M. Waser, Eur. J. Org. Chem., 2013, 637–648 CrossRef CAS; (c) R. Herchl and M. Waser, Tetrahedron Lett., 2013, 54, 2472 CrossRef CAS PubMed; (d) K. M. Johnson, M. S. Rattley, F. Sladojevich, D. M. Barber, M. G. Nuñez, A. M. Goldys and D. J. Dixon, Org. Lett., 2012, 14, 2492 CrossRef CAS PubMed; (e) C. D. Fiandra, L. Piras, F. Fini, P. Disetti, M. Moccia and M. F. A. Adamo, Chem. Commun., 2012, 48, 3863 RSC; (f) P. Elsner, L. Bernardi, G. Dela Salla, J. Overgaard and K. A. Jørgensen, J. Am. Chem. Soc., 2008, 130, 4897 CrossRef CAS PubMed.
- (a) S. E. Denmark, N. D. Gould and L. M. Wolf, J. Org. Chem., 2011, 76, 4260 CrossRef CAS PubMed; (b) S. E. Denmark, N. D. Gould and L. M. Wolf, J. Org. Chem., 2011, 76, 4337 CrossRef CAS PubMed; (c) L. Wang, S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2011, 50, 5327 CrossRef CAS PubMed; (d) S. Shirakawa and K. Maruoka, Tetrahedron Lett., 2014, 55, 3833 CrossRef CAS; (e) G. P. Petrova, H.-B. Li, K. Maruoka and K. Morokuma, J. Phys. Chem. B, 2014, 118, 5154 CrossRef CAS PubMed.
- This screening should not be considered exhaustive, since, for brevity, we reported only the most significant experiments.
- (a) A. Berkessel, M. Guixà, F. Schmidt, J. M. Neudörfl and J. Lex, Chem.–Eur. J., 2007, 13, 4483 CrossRef CAS PubMed; (b) H.-J. Lee and C.-W. Cho, J. Org. Chem., 2015, 80, 11435 CrossRef CAS PubMed.
- For a review on bifunctional chiral ammonium salts and leading references see: J. Novacek and M. Waser, Eur. J. Org. Chem., 2014, 802–809 CrossRef CAS PubMed.
- For a book and selected examples see: (a) Enantiomer Separation: Fundamentals and Practical Methods, ed. Fumio Toda, Kluwer Academic Publishers, Â Dordrecht, Boston, London, 2004 Search PubMed; (b) W.-W. Chen, M.-H. Xu and G.-Q. Lin, Tetrahedron Lett., 2007, 48, 7508 CrossRef CAS; (c) A. Ishii, M. Kanai, K. Higashiyama and k. Mikami, Chirality, 2002, 14, 709 CrossRef CAS PubMed.
- (a) G. K. Surya Prakash, F. Wang, C. Ni, J. Shen, R. Haiges, A. K. Yudin, T. Mathew and G. A. Olah, J. Am. Chem. Soc., 2011, 133, 9992 CrossRef PubMed; (b) G. K. Surya Prakash, F. Wang, M. Rahm, Z. Zhang, C. Ni, J. Shen and G. A. Olah, J. Am. Chem. Soc., 2014, 136, 10418 CrossRef PubMed.
- G. P. Petrova, H.-B. Li, K. Maruoka and K. Morokuma, J. Phys. Chem. B, 2014, 118, 5154 CrossRef CAS PubMed.
- (a) D. Uraguchi, K. Koshimoto and T. Ooi, J. Am. Chem. Soc., 2008, 130, 10878 CrossRef CAS PubMed; (b) D. Uraguchi, K. Oyaizu and T. Ooi, Chem.–Eur. J., 2012, 18, 8306 CrossRef CAS PubMed.
- Q. Wang, Q. Wang, B. Zhang, X. Sun and S. Zhang, Synlett, 2009, 1311 Search PubMed.
- B. A. Provencher, K. J. Bartelson, Y. Liu, B. M. Foxman and L. Deng, Angew. Chem., Int. Ed., 2011, 50, 10565 CrossRef CAS PubMed.
- H. Li, Y. Wang, L. Tang and L. Deng, J. Am. Chem. Soc., 2004, 126, 9906 CrossRef CAS PubMed.
- Spartan’04, Wavefunction, Inc., Irvine, CA Search PubMed.
- S. Schenker, C. Schneider, S. B. Tsogoeva and T. Clark, J. Chem. Theory Comput., 2011, 7, 3586 CrossRef CAS PubMed.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef.
- J. Cioslowski, J. Am. Chem. Soc., 1989, 111, 8333 CrossRef CAS.
- M. J. Frisch, et al. Gaussian 09 Revision D.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- (a) A. Russo, A. Capobianco, A. Perfetto, A. Lattanzi and A. Peluso, Eur. J. Org. Chem., 2011, 1922 CrossRef CAS; (b) A. Capobianco, A. Russo, A. Lattanzi and A. Peluso, Adv. Synth. Catal., 2012, 354, 2789 CrossRef CAS; (c) T. Caruso, A. Capobianco and A. Peluso, J. Am. Chem. Soc., 2007, 129, 15347 CrossRef CAS PubMed.
- S. Meninno, A. Capobianco, A. Peluso and A. Lattanzi, Green Chem., 2015, 17, 2317 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra05488a |
| This journal is © The Royal Society of Chemistry 2016 |