
Diketopyrrolopyrrole-based polymer with a semi-fluorinated side chain for high-performance organic thin-film transistors†
Abstract
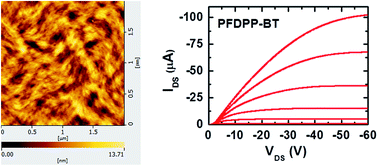
A diketopyrrolopyrrole (DPP) unit with a semi-fluorinated side chain was copolymerized with bithiophenyl (BT) to afford a novel donor–acceptor (D–A) conjugated polymer PFDPP–BT (FDPP refers to the semi-fluorinated DPP unit). The non-fluorinated polymer PDPP–BT was synthesized as the reference polymer to investigate the effect of fluorination of side chains on the morphology of thin films and charge-carrier mobility. Organic thin-film transistors based on these two polymers showed decent device performance. Interestingly, the semiconductor polymer PFDPP–BT exhibited hole mobility about 3 times higher than that of the related PDPP–BT. Atomic force microscopy (AFM) and X-ray diffraction (XRD) measurements demonstrated that the semi-fluorinated side chains had a great effect on the thin film morphology and crystallinity, which led to enhancement in device performance.
 Please wait while we load your content...
Please wait while we load your content...

