
Silicon direct bonding via low-temperature wet chemical surface activation
Abstract
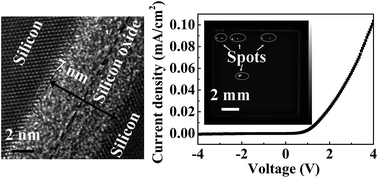
Commercial silicon substrates were bonded via low-temperature wet chemical surface activation in a standard laboratory without ultrahigh vacuum or a clean room. A smooth direct bond with no voids or microcracks was obtained, and the best bonding strength reached up to 4.15 MPa at 250 °C with the 25 MPa pressure-assisted process. The p–n junction prepared using this method exhibited excellent I–V characteristics. An updated model for the silicon direct bonding mechanism was proposed and proved. Raman scattering analysis indicated an increase in the Si–OH density on the silicon surface after the activation treatment. Cross-sectional HRTEM and EELS analysis crossing the interface indicated that the Si–O–Si linkage on the bonding interface that formed after low-temperature annealing was of a lower density than that of the native oxide. The thickness of the bonding interface decreased and the silicate linkage underwent further densification when high-temperature annealing was executed.
 Please wait while we load your content...
Please wait while we load your content...

