
Experimental and theoretical approach to account for green luminescence from Gd2Zr2O7 pyrochlore: exploring the site occupancy and origin of host-dopant energy transfer in Gd2Zr2O7:Eu3+
Abstract
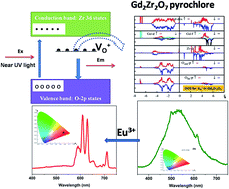
Pure and Eu3+-doped Gd2Zr2O7 pyrochlore was synthesized using a gel combustion method using citric acid as a fuel. Samples of Gd2Zr2O7 pyrochlore were characterized systematically using X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FTIR), scanning electron microscopy (SEM), energy-dispersive spectroscopy (EDS) and time-resolved photoluminescence spectroscopy (TRPLS). On irradiating the undoped Gd2Zr2O7 pyrochlore with Ultraviolet (UV) light, an intense green emission was observed. Photoluminescence lifetime measurements and X-ray photoelectron spectroscopy (XPS) showed the presence of oxygen vacancies in the pyrochlore sample, which were responsible for the intense green emission in Gd2Zr2O7. Calculations based on density functional theory (DFT+U) of the electronic density of states in the presence of charged oxygen defects qualitatively explained the origin of the green emission in undoped Gd2Zr2O7. The emission spectrum of Eu3+ revealed that it was distributed at both Gd3+ and Zr4+ sites in Gd2Zr2O7, which was also confirmed using lifetime measurements. DFT-based calculations of cohesive energy also showed that doping with Eu is almost equally favorable at Gd and Zr sites. Based on DFT calculations, it is proposed that the distribution of f and d states of Eu3+ atoms matches well with the total density of states (DOS) of ordered Gd2Zr2O7 (o-Gd2Zr2O7), which signifies efficient transfer of photon energy from the host to the Eu3+ dopant. The actual site symmetry of europium ions in gadolinium zirconate was also determined based on the Stark splitting pattern and was found to be D2d, although it is Oh for Gd3+ in Gd2Zr2O7. Calculations of the Judd–Ofelt parameters revealed that Ω2 > Ω4, which indicates high covalency and low symmetry around Eu3+, which is in agreement with the results of emission spectroscopy. The high intensity of the red emission corresponding to the 5D0 → 7F2 transition and good fluorescence yields (51%) highlight the unexplored potential of Gd2Zr2O7:Eu3+ as a promising red phosphor.
 Please wait while we load your content...
Please wait while we load your content...

