
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of hollow PtAg alloy nanospheres with excellent electrocatalytic performances towards methanol and formic acid oxidations†
Fulin
Zheng
,
Sin-Yee
Luk
,
Tsz-Lung
Kwong
 and
Ka-Fu
Yung
*
and
Ka-Fu
Yung
*
Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong. E-mail: bckfyung@polyu.edu.hk; Fax: +852 2364 9932; Tel: +852 3400 8863
First published on 26th April 2016
Abstract
Hollow PtAg alloy nanospheres were synthesized via galvanic replacement reaction between silver nanoparticles and K2PtCl4 at 60 °C. Their morphology, composition and structure were characterized by UV-vis, XRD, TEM, HRTEM, SAED, EDX and XPS. It was found that the shell thickness and roughness could be controlled by varying the amount of Pt precursor solutions. Compared with commercially available Pt black, the PtAg showed higher catalytic activity and higher CO tolerance towards the electro-oxidations of methanol and formic acid, which make them a prominent catalyst for DMFCs and DFAFCs.
Owing to the increasing demands for energy and growing concerns about global warming and air pollution, intensive growth in the research of fuel cells has been stimulated.1,2 Direct methanol fuel cells (DMFCs) and direct formic acid fuel cells (DFAFCs) have attracted growing attention as energy conversion devices for portable applications. Both methanol and formic acid have low cost, high abundance, low toxicity and can be transported and stored safely when compared with hydrogen as a fuel.3,4 In DMFCs and DFAFCs, platinum-based electrocatalysts are used as anode catalysts. However, platinum (Pt) is a rare and precious metal. It is also easily deteriorated by COads poisoning effect during the oxidation of methanol and formic acid.5 Hence, the massive application is constrained by its limited availability and severe deterioration during the oxidation of fuels. Therefore, it is crucial to investigate and explore on the development of the Pt-based electrocatalysts of improved Pt utilization and enhanced electrocatalytic activity and stability in fuel cells.
To achieve this purpose, Pt-based electrocatalysts are significantly investigated including Pt-alloy catalysts,6–9 core–shell structured Pt-based catalysts,10–13 Pt-monolayer catalysts,12,14,15 Pt-based catalysts with high index,16 nanoporous Pt-based catalysts17,18 and hollow Pt-based nanocatalysts.19,20 From these examples, it is found that Pt-based catalysts with high index planes, which are associated with atomic steps, edges and kinks, usually exhibit the enhanced electrocatalytic activity and stability.16,21 In the recent years, hollow Pt-based alloy nanostructures with nanoporosity have drawn much more attention due to their large electrochemical active surface area (ECSA) resulting in higher catalytic activity and improved utilization of precious Pt metal.22,23
Lee et al.24 reported the synthesis of hollow and nanoporous PtAu alloy nanoparticles by galvanic replacement reaction using silver (Ag) nanoparticles as the templates and a high electrocatalytic activity was exhibited towards the oxidation of formic acid. To further reduce the cost of the catalysts, Ag has also been investigated to be incorporated into Pt metal, instead of gold (Au), due to its lower cost.25 Hollow PtAg nanostructures with controlled number of voids including nanoboxes, dimers, multimers and popcorn-shaped nanostructures were synthesized by galvanic replacement reaction with the existence of hydrochloric acid (HCl). These hollow PtAg nanostructures exhibited large surface area and high catalytic activity towards the oxidation of methanol.26 Xu et al.27 successfully prepared metallic PtAg alloy nanoparticles supported on carbon powder by co-reduction method. The as-prepared PtAg alloy catalysts showed better catalytic activity than commercial carbon-supported Pt towards the oxidation of formic acid.
In this study, hollow PtAg alloy nanospheres were synthesized by galvanic replacement reaction between Ag nanoparticles and K2PtCl4 at 60 °C. The morphology, crystal structure and surface composition of the synthesized hollow PtAg nanospheres were characterized by various techniques including TEM, EDX, SAED, XRD and XPS analysis. Besides, the electrocatalytic activity and stability of the catalysts were examined and compared with Pt black towards the oxidations of methanol and formic acid.
UV-visible spectrometry is a commonly used technique to study the Ag-related chemistry, due to the surface plasmon resonance (SPR) effect, a characteristic absorption peak of Ag nanoparticles can be easily obtained.28Fig. 1 shows the spectra of a series of pure Ag nanoparticles and PtAg nanospheres prepared by the addition of different volumes of 25 mM K2PtCl4 solution (Table 1). The pure Ag nanoparticles show a characteristic SPR peak at 395 nm and as the amount of the Pt precursor increases, the intensity of the Ag absorption peak decreases suggesting a gradual increase of the deposition of Pt atoms on the surface of Ag nanoparticles.29 When the amount of K2PtCl4 solution was increased to 550 μL, a flat line is achieved indicating the disappearance of the silver core. The gradual decrease of the Ag-related absorbance peak may imply the formation of a hollow structure.
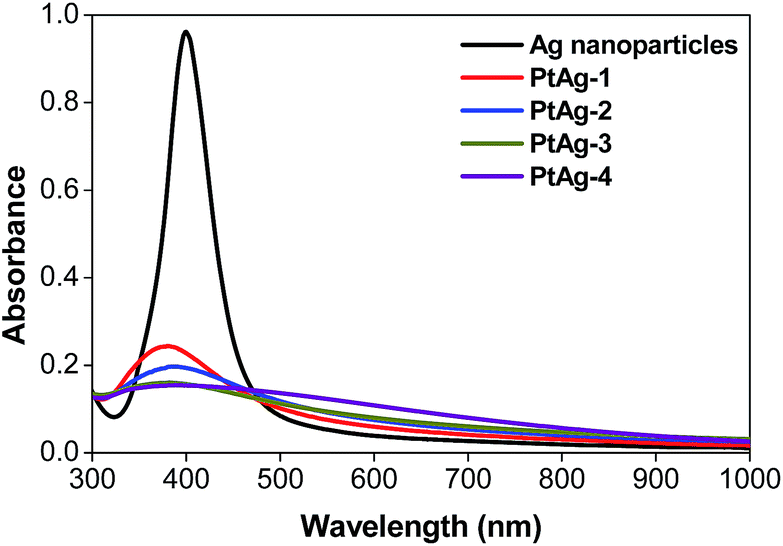 | ||
| Fig. 1 UV-vis spectra of the Ag nanoparticles and a series of PtAg nanospheres. | ||
| PtAg nanosphere | Volume of 25 mM K2PtCl4 solution (μL) |
|---|---|
| PtAg-1 | 150 |
| PtAg-2 | 250 |
| PtAg-3 | 350 |
| PtAg-4 | 550 |
The morphology of Ag templates was characterized by TEM analysis as shown in Fig. 2. The Ag templates were in spherical morphology with a diameter ranging from 20 to 50 nm. To analyze crystal structure, SAED analysis was performed with the result displayed in Fig. 2(b). The diffraction rings are assigned to the (111), (200), (220) and (311) crystal planes which align with the d-spacing of Ag. It suggests that the Ag nanoparticles adopt a face-centered cubic (fcc) crystal system. As depicted in Fig. 2(c), the diffraction fringe with d-spacing of 0.24 nm is corresponded to the (111) crystal plane of Ag.
 | ||
| Fig. 2 (a) TEM micrograph, (b) SAED analysis and (c) HR-TEM micrograph of Ag nanoparticles. | ||
From the UV-vis analysis, it is proposed that the amount of Pt atoms deposited on the surface of the Ag nanoparticles increases with an increase in the volume of K2PtCl4 solution added to the Ag colloidal solution. TEM analysis (Fig. 3) was carried out to further confirm this proposal. Hollow PtAg nanospheres were successfully synthesized due to galvanic replacement reaction. With an increase in the amount of K2PtCl4 solution, more hollow nanostructures were observed. The galvanic replacement reaction between Ag and K2PtCl4 is as follow,26
| 2Ag + K2PtCl4 → Pt + 2AgCl + 2KCl | (1) |
 | ||
| Fig. 3 TEM micrographs of hollow (a) PtAg-1, (b) PtAg-2, (c) PtAg-3, (d) PtAg-4 and (e) EDX spectrum of hollow PtAg-4 nanospheres before washing. | ||
The active Ag atoms first react with Pt(II) ions to yield Pt atoms which deposited on to the surface of the Ag templates forming pinholes. As the reaction proceeded, hollow nanostructures were formed due to dissolution of Ag from the pinholes and continuous deposition of Pt atoms onto the surface.
Elemental analysis of all hollow PtAg nanospheres showed similar EDX spectrum (Fig. 3(e)). It displays the signal for chlorine apart from silver and platinum. It is proposed that the remaining solid cores at low amount of K2PtCl4 are the unreacted Ag and silver chloride (AgCl).30 During the galvanic replacement reaction, AgCl was the major by-product which contaminated the hollow PtAg nanospheres.26 Although AgCl would dissolve at relatively high temperature,31 chlorine is still detected. In order to remove AgCl and unreacted Ag, hollow PtAg nanospheres were washed with 35% NH4OH solution and 5 M HNO3 solution respectively.
Washed hollow PtAg nanospheres were characterized by TEM analysis. As shown in Fig. 4, the roughness of the shell surface increases with an increase in the amount of K2PtCl4 solution. The shell surface exhibits as a single smooth layer when lower amount of K2PtCl4 solution is added while the shell surface becomes rougher with the development of aggregated nanoparticles on the shell surface when higher amount of K2PtCl4 solution is added. As discussed, Pt atoms were formed on the surface of Ag nanoparticles and the adsorbed AgCl. During galvanic replacement reaction, Ag atoms migrate from the core and diffuse through the attached AgCl to the surface of Ag nanoparticles to react with K2PtCl4 to form alloy or AgCl. Higher roughness of the nanostructures would be formed due to formation of more AgCl with higher amount of K2PtCl4 solution was added.26 The relationship between the thickness of hollow PtAg nanospheres and the different amount of K2PtCl4 solution is clearly shown in Table 2. With an increase in the amount of K2PtCl4 from 150 μL to 550 μL, the thickness increases from 2.3 nm to 7.5 nm.
 | ||
| Fig. 4 TEM micrographs of hollow (a) PtAg-1, (b) PtAg-2, (c) PtAg-3 and (d) PtAg-4, (e) HR-TEM micrograph and (f) SAED analysis on washed hollow PtAg-4 nanosphere. | ||
| PtAg nanosphere | Shell thickness (nm) | Theoretical Pt/Ag | Pt/Ag from EDX |
|---|---|---|---|
| PtAg-1 | 2.3 | 0.28 | 7.25 |
| PtAg-2 | 3.6 | 0.46 | 6.76 |
| PtAg-3 | 5.1 | 0.83 | 5.27 |
| PtAg-4 | 7.5 | 1.02 | 4.77 |
EDX analysis (Table 2) on washed hollow PtAg nanospheres suggests a decreasing trend of the atomic Pt-to-Ag ratio with an increase of the amount of K2PtCl4 solution. With the addition of K2PtCl4 solution, more AgCl were formed and adsorbed on the surface of Ag nanoparticle. Since AgCl acts as a secondary template for the galvanic replacement reaction with K2PtCl4 to form alloy, hollow PtAg nanospheres with higher content of Ag would be resulted.26
All washed hollow PtAg nanospheres demonstrated similar XRD pattern (Fig. 5). Several diffraction peaks located at 39.1°, 45.7°, 67.1° and 80.1° are assigned to be (111), (200), (220) and (311) crystal planes of fcc structure respectively.32 As summarized in Table 3, the diffraction peaks of PtAg are located in between standard diffraction peaks of Ag and Pt and no characteristic Ag or Pt diffraction peaks are found, implying the formation of PtAg alloy in the galvanic replacement reaction.33
 | ||
| Fig. 5 XRD pattern of washed hollow PtAg-4 nanospheres. | ||
| Crystal plane | Pt (°) | Ag (°) | PtAg (°) |
|---|---|---|---|
| (111) | 39.7 | 38.0 | 39.1 |
| (200) | 46.3 | 44.3 | 45.7 |
| (222) | 68.3 | 64.8 | 67.1 |
| (311) | 81.2 | 77.5 | 80.1 |
HR-TEM analysis on washed hollow PtAg nanospheres (Fig. 4(e)) shows several diffraction fringes with d-spacing of 2.29 Å, 2.30 Å, 2.31 Å which correspond to (111) crystal plane of fcc structure.34 It is found that the d-spacing of diffraction fringes in PtAg are located in between the d-spacing of crystal plane of Pt (2.26 Å) and Ag (2.35 Å).35 This result further confirms that the formation of PtAg alloy due to the galvanic displacement reaction. SAED analysis (Fig. 4(f)) shows the diffraction rings of (111), (200), (220) and (311) crystal planes which is consistent to the results obtained from XRD analysis.
XPS analysis, as illustrated in Fig. 6, was conducted to further investigate the surface composition of washed hollow PtAg nanospheres and the interaction between Pt and Ag. The peak of Pt 4f and Ag 3d shift to higher and lower binding energy respectively as compared with pure Pt and Ag metal. It suggests that the electron transfer from Pt to Ag related to electronic interaction between Ag and Pt and further confirms the formation of PtAg alloy.27,36,37
 | ||
| Fig. 6 Survey XPS spectra of (a) Pt 4f and (b) Ag 3d of washed hollow PtAg-4 nanospheres. The dotted line represents the binding energy of pure Pt and Ag metal respectively. | ||
The electrocatalytic activity of the PtAg alloy was characterized by CV as shown in Fig. 7(a). The CVs of PtAg alloy and commercially available Pt black exhibit hydrogen adsorption and desorption peak in the potential range from 0.24 V to 0.05 V. The Pt black demonstrates two resolved peaks in hydrogen adsorption and desorption region respectively. This was associated with weakly and strongly bonded hydrogen species on different crystal planes of Pt.35,38 The peaks with more negative potential and those with more positive potential correspond to (111) plane and (100) plane respectively.39,40 However, no resolved peaks are observed in the CV of PtAg alloy which could be attributed to the incorporation of Ag into the Pt lattice, decreasing the crystallinity of the surface. For PtAg alloy, an anodic peak is observed at 0.8 V to 1.0 V due to the formation of the oxide species of Ag and Pt.27 In contrast, only an anodic peak for oxidation of Pt is observed in Pt black at around 0.6 V. Both catalysts exhibit the reduction of Pt oxides at around 0.4 V to 0.5 V in cathodic scan.
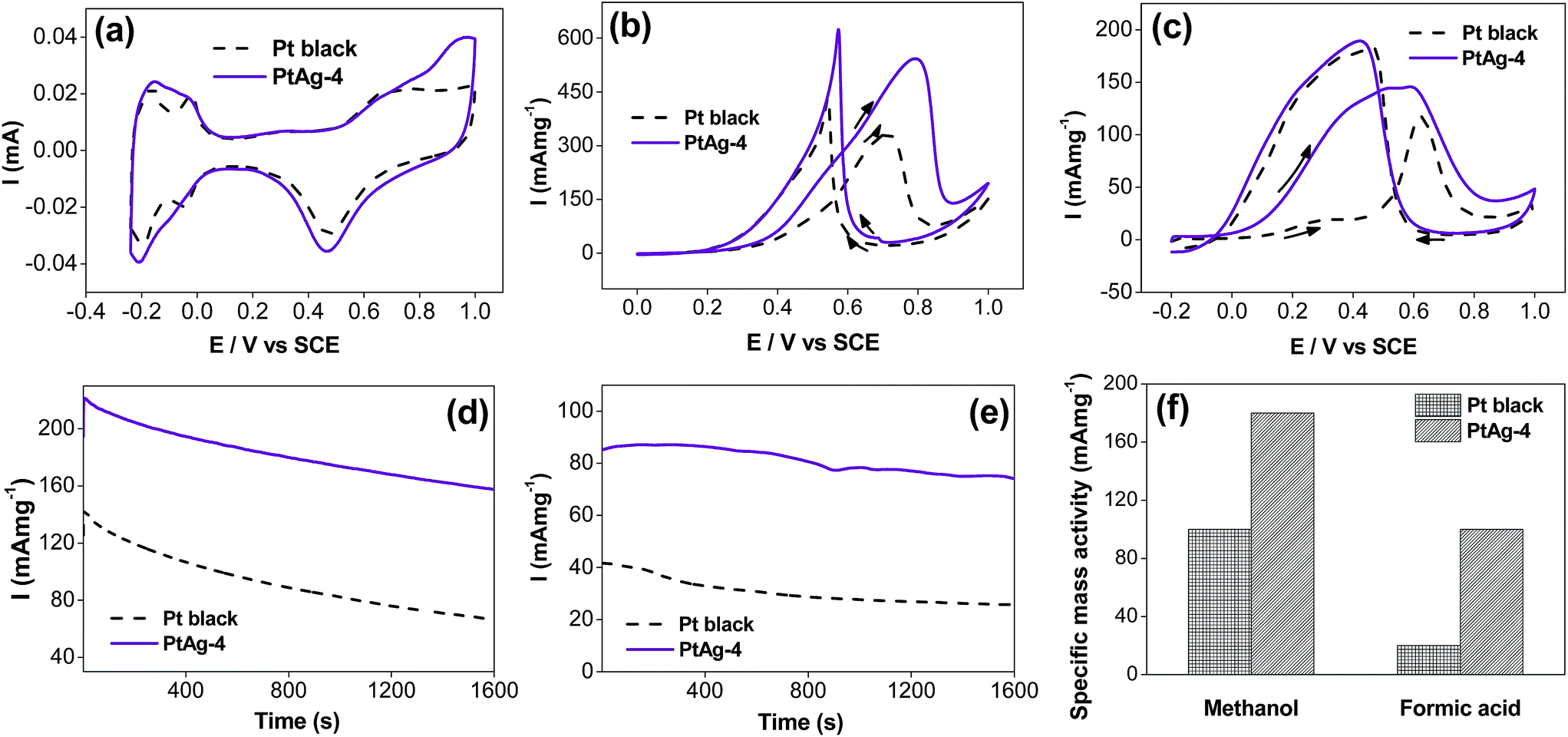 | ||
| Fig. 7 CVs of PtAg-4 nanosphere and Pt black in (a) N2-purged 0.1 M H2SO4 solution, (b) 0.1 M methanol + 0.1 M H2SO4 and (c) 0.05 M HCOOH + 0.1 M H2SO4 solutions with a scan rate of 50 mV s−1. Chronoamperometric tests of PtAg-4 nanosphere and Pt black in (d) 0.1 M methanol + 0.1 M H2SO4 and (e) 0.05 M HCOOH + 0.1 M H2SO4 solutions at fixed potentials of 0.5 V and 0.3 V respectively with a scan rate of 50 mV s−1. (f) Specific mass activity of PtAg-4 nanosphere and Pt black in methanol and formic acid oxidations. | ||
The ECSA of the catalysts can be calculated by the following equation,41,42
| ECSA = QH/mqH | (2) |
The ECSA for PtAg alloy and Pt black are calculated to be 18.7 m2 g−1 and 14.9 m2 g−1 respectively. On the other hand, the larger ECSA of the hydrogen adsorption and desorption peaks of PtAg alloy indicates better utilization of Pt metal for hollow nanostructure catalysts.
The electrocatalytic activities of PtAg alloy Pt black towards methanol and formic acid oxidations were investigated. For the electro-oxidation of methanol as displayed in Fig. 7(b), anodic peaks at 0.7 V to 0.8 V in forward scan and at 0.5 V to 0.6 V in the backward scan are observed. It was found that the specific activity towards methanol oxidation at 0.6 V decreases with a decrease in the atomic Pt-to-Ag ratio (see ESI section Fig. S1†). The increasing Ag content in the hollow PtAg nanospheres and increasing shell thickness may decrease the accessibility of methanol to PtAg surface.25Fig. 7(f) clearly summarizes that the PtAg alloy shows 1.8 times higher specific mass activity than Pt black at 0.5 V.
For the electro-oxidation of formic acid as shown in Fig. 7(c), the onset potential of PtAg alloy is 40 mV negatively shifted when compared with Pt black, suggesting easier oxidation of formic acid. A broad peak observed in forward scan of PtAg alloy and a shoulder peak at 0.2 V to 0.4 V in Pt black indicate the dehydrogenation of formic acid, however, an exceptional peak at 0.62 V in Pt black attributes to the indirect oxidation of formic acid. It is well-known that the formic acid oxidation catalysed by Pt can go through direct and indirect oxidation pathways which result in distinct oxidation peak in the forward scan at 0.3 V and 0.6 V respectively.12 The specific activity towards the formic acid oxidation decreases with an increase in the amount of K2PtCl4 from 150 μL to 250 μL. The specific activity towards the formic acid oxidation increase when the amount of K2PtCl4 increases from 250 μL to 550 μL (see ESI section Fig. S2†). It is proposed that the smooth and porous shell and high atomic Pt-to-Ag ratio of PtAg-1 increase the accessibility of formic acid to PtAg surface, leading the highest specific activity in both direct and indirect oxidation of formic acid. PtAg-2 exhibits the lowest specific activity towards direct and indirect oxidation of formic acid due to the aggregation of nanoparticles on the shell surface which decreases the accessible surface area. Afterwards, the increase of specific activity towards direct and indirect oxidation of formic acid of PtAg-3 and PtAg-4 are observed due to the formation of more nanoparticles on the shell surface which expose more active surface area. Also, it is proposed that the existence of Ag may eliminate the formation of intermediate which would enhance the specific activity towards the formic acid oxidation.43,44 At 0.3 V, PtAg alloy shows 5 times higher specific mass activity than Pt black, as displayed in Fig. 7(f), which suggests that less poisoning CO-like intermediate is formed and adsorbed on the surface of PtAg alloy. It is proposed that the incorporation of Ag increases the 5d-orbitals vacancy of Pt, reducing back-donation of 5d electrons to CO, and thus weakening the interaction between Pt and COads and enhancing the electrocatalytic capability.36,45–47
To study the electrocatalytic stability of the PtAg alloy and Pt black towards the oxidations of methanol and formic acid, chronoamperometric tests were conducted for 1600 s under room temperature and the results are shown in Fig. 7(d and e). Both specific mass activities in the chronoamperometric tests decrease after 1600 s. At a fixed potential, intermediate species are formed due to the continuous oxidation of the fuels.48 The accumulation and adsorption of intermediate species on the surface of electro-catalysts block the active sites and refrain from the oxidation of fuels.49 It is found that the stability of PtAg alloy is higher than that of Pt black. The specific mass activities of PtAg alloy remains 81.4% and 87.2% while those of Pt black remains 44.8% and 61.1% towards the oxidations of methanol and formic acid respectively. These results relate to the better CO tolerance of PtAg alloy due to the electronic effect caused by the incorporation of Ag.
Conclusions
In summary, hollow PtAg alloy nanospheres were successfully synthesized by galvanic replacement reaction between Ag nanoparticles and K2PtCl4 at 60 °C. The shell thickness and the atomic ratio of Pt/Ag of the hollow nanospheres could be controlled by varying the volumes of Pt precursor solution. The as-synthesized hollow PtAg alloy nanospheres showed higher electrocatalytic activity and stability towards methanol and formic acid oxidations than commercially available Pt black and will be promising electro-catalysts for DMFCs and DFAFCs.Acknowledgements
The authors gratefully acknowledge the financial support from The Hong Kong Research Grants Council (PolyU5029/12P) and The Hong Kong Polytechnic University. S. Y. Luk acknowledges the receipt of postgraduate studentship administrated by The Hong Kong Polytechnic University.Notes and references
- A. M. Abdullah, A. M. Mohammad, T. Okajima, F. Kitamura and T. Ohsaka, J. Power Sources, 2009, 190, 264–270 CrossRef CAS.
- A. M. Abdullah, T. Okajima, A. M. Mohammad, F. Kitamura and T. Ohsaka, J. Power Sources, 2007, 172, 209–214 CrossRef CAS.
- Y.-Y. Chu, Z.-B. Wang, Z.-Z. Jiang, D.-M. Gu and G.-P. Yin, J. Power Sources, 2012, 203, 17–25 CrossRef CAS.
- A. Kirubakaran, S. Jain and R. K. Nema, Renewable Sustainable Energy Rev., 2009, 13, 2430–2440 CrossRef CAS.
- Y. Zhang, H. Shu, G. Chang, K. Ji, M. Oyama, X. Liu and Y. He, Electrochim. Acta, 2013, 109, 570–576 CrossRef CAS.
- J.-H. Choi, K.-W. Park, I.-S. Park, W.-H. Nam and Y.-E. Sung, Electrochim. Acta, 2004, 50, 787–790 CrossRef CAS.
- K. Wang, H. A. Gasteiger, N. M. Markovic and P. N. Ross Jr, Electrochim. Acta, 1996, 41, 2587–2593 CrossRef CAS.
- W. Chen, J. Kim, S. Sun and S. Chen, Langmuir, 2007, 23, 11303–11310 CrossRef CAS PubMed.
- C. Wang, N. M. Markovic and V. R. Stamenkovic, ACS Catal., 2012, 2, 891–898 CrossRef CAS.
- W. He, X. Wu, J. Liu, K. Zhang, W. Chu, L. Feng, X. Hu, W. Zhou and S. Xie, Langmuir, 2009, 26, 4443–4448 CrossRef PubMed.
- S. Wojtysiak, J. Solla-Gullón, P. Dłużewski and A. Kudelski, Colloids Surf., A, 2014, 441, 178–183 CrossRef CAS.
- F. Zheng, W.-T. Wong and K.-F. Yung, Nano Res., 2014, 7, 410–417 CrossRef CAS.
- H. Zhang, C. Cao, J. Zhao, R. Lin and J. Ma, Chin. J. Catal., 2012, 33, 222–229 CAS.
- Y. Ando, K. Sasaki and R. Adzic, Electrochem. Commun., 2009, 11, 1135–1138 CrossRef CAS.
- N. Kristian, Y. Yan and X. Wang, Chem. Commun., 2008, 353–355, 10.1039/b714230g.
- N. Tian, Z.-Y. Zhou and S.-G. Sun, J. Phys. Chem. C, 2008, 112, 19801–19817 CAS.
- Z. Zhang, Y. Wang and X. Wang, Nanoscale, 2011, 3, 1663–1674 RSC.
- J. Wang, P. Holt-Hindle, D. MacDonald, D. F. Thomas and A. Chen, Electrochim. Acta, 2008, 53, 6944–6952 CrossRef CAS.
- H.-P. Liang, H.-M. Zhang, J.-S. Hu, Y.-G. Guo, L.-J. Wan and C.-L. Bai, Angew. Chem., 2004, 116, 1566–1569 CrossRef.
- J. Zhao, W. Chen, Y. Zheng and X. Li, J. Power Sources, 2006, 162, 168–172 CrossRef CAS.
- T. Yu, D. Y. Kim, H. Zhang and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 2773–2777 CrossRef CAS PubMed.
- E. Anumol, A. Halder, C. Nethravathi, B. Viswanath and N. Ravishankar, J. Mater. Chem., 2011, 21, 8721–8726 RSC.
- X. Zhou, Y. Gan, J. Du, D. Tian, R. Zhang, C. Yang and Z. Dai, J. Power Sources, 2013, 232, 310–322 CrossRef CAS.
- D. Lee, H. Y. Jang, S. Hong and S. Park, J. Colloid Interface Sci., 2012, 388, 74–79 CrossRef CAS PubMed.
- C. Li and Y. Yamauchi, Phys. Chem. Chem. Phys., 2013, 15, 3490–3496 RSC.
- W. Zhang, J. Yang and X. Lu, ACS Nano, 2012, 6, 7397–7405 CrossRef CAS PubMed.
- J. B. Xu, T. S. Zhao and Z. X. Liang, J. Phys. Chem. C, 2008, 112, 17362–17367 CAS.
- J. Zeng, Y. Zheng, M. Rycenga, J. Tao, Z.-Y. Li, Q. Zhang, Y. Zhu and Y. Xia, J. Am. Chem. Soc., 2010, 132, 8552–8553 CrossRef CAS PubMed.
- B. Y. A. B. Q. X. D. Zhao, Electrochem. Commun., 2008, 10, 884–887 CrossRef.
- J. Yang, J. Y. Lee, H.-P. Too and S. Valiyaveettil, J. Phys. Chem. B, 2006, 110, 125–129 CrossRef CAS PubMed.
- Y. Sun and Y. Xia, J. Am. Chem. Soc., 2004, 126, 3892–3901 CrossRef CAS PubMed.
- J. Xu, K. Hua, G. Sun, C. Wang, X. Lv and Y. Wang, Electrochem. Commun., 2006, 8, 982–986 CrossRef CAS.
- Z. Peng and H. Yang, J. Solid State Chem., 2008, 181, 1546–1551 CrossRef CAS.
- Y.-Y. Feng, J.-H. Ma, G.-R. Zhang, G. Liu and B.-Q. Xu, Electrochem. Commun., 2010, 12, 1191–1194 CrossRef CAS.
- S. Yu, Q. Lou, K. Han, Z. Wang and H. Zhu, Int. J. Hydrogen Energy, 2012, 37, 13365–13370 CrossRef CAS.
- S. Schwamborn, M. Bron and W. Schuhmann, Electroanalysis, 2011, 23, 588–594 CAS.
- Y.-Y. Feng, G.-R. Zhang, J.-H. Ma, G. Liu and B.-Q. Xu, Phys. Chem. Chem. Phys., 2011, 13, 3863–3872 RSC.
- J. Zhao and A. Manthiram, Appl. Catal., B, 2011, 101, 660–668 CrossRef CAS.
- P. N. Ross Jr, Surf. Sci., 1981, 102, 463–485 CrossRef.
- N. M. Marković and P. N. Ross Jr, Surf. Sci. Rep., 2002, 45, 117–229 CrossRef.
- T. J. Schmidt, H. A. Gasteiger, G. D. Stäb, P. M. Urban, D. M. Kolb and R. J. Behm, J. Electrochem. Soc., 1998, 145, 2354–2358 CrossRef CAS.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- J. Xu, T. Zhao and Z. Liang, J. Phys. Chem. C, 2008, 112, 17362–17367 CAS.
- X. Cao, N. Wang, Y. Han, C. Gao, Y. Xu, M. Li and Y. Shao, Nano Energy, 2015, 12, 105–114 CrossRef CAS.
- L. G. S. Pereira, V. A. Paganin and E. A. Ticianelli, Electrochim. Acta, 2009, 54, 1992–1998 CrossRef CAS.
- H. Igarashi, T. Fujino, Y. Zhu, H. Uchida and M. Watanabe, Phys. Chem. Chem. Phys., 2001, 3, 306–314 RSC.
- Y.-Y. Feng, L.-X. Bi, Z.-H. Liu, D.-S. Kong and Z.-Y. Yu, J. Catal., 2012, 290, 18–25 CrossRef CAS.
- B. Liu, J. H. Chen, C. H. Xiao, K. Z. Cui, L. Yang, H. L. Pang and Y. F. Kuang, Energy Fuels, 2007, 21, 1365–1369 CrossRef CAS.
- T. Xia, H. Shen, G. Chang, Y. Zhang, H. Shu, M. Oyama and Y. He, J. Nanomater., 2014, 2014, 7 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra06398e |
| This journal is © The Royal Society of Chemistry 2016 |