
Layered NiO/reduced graphene oxide composites by heterogeneous assembly with enhanced performance as high-performance asymmetric supercapacitor cathode†
Qian Li‡
ab,
Qiang Wei‡c,
Lijing Xie*d,
Chengmeng Chen*d,
Chunxiang Lu*a,
Fang-Yuan Sud and
Pucha Zhoua
aNational Engineering Laboratory for Carbon Fiber Technology, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, China. E-mail: lucx@sxicc.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cSchool of Chemical & Biological Engineering, Lanzhou Jiaotong University, Lanzhou 730070, China
dKey Laboratory of Carbon Materials, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, China. E-mail: ccm@sxicc.ac.cn
First published on 6th May 2016
Abstract
A layered NiO/reduced graphene oxide composite (NiO/RGO) was prepared by a facile heterogeneous assembly approach with subsequent in situ thermal reduction. The two-dimensional hydroxide and graphene oxide nanosheets achieved nanoscale dispersion in the composites, with high interfacial interaction between each other. The as-obtained material exhibits a high specific capacitance of 782 F g−1 at 0.5 A g−1 and an excellent cycling stability with 94.1% retention at 2 A g−1 after 3000 cycles in a three-electrode system. To further evaluate the NiO/RGO electrode for practical application, an asymmetric supercapacitor NiO/RGO//AC was fabricated using the NiO/RGO as cathode and AC as anode. It exhibits maximum energy density of 32.5 W h kg−1 at a power density of 375 W kg−1 and even retains 19.78 W h kg−1 at 7500 W kg−1. This asymmetric device also shows a high cycling stability along with 92.7% retention after 3000 cycles, and is able to light up an LED bulb. The success of the NiO/RGO composite sheds light on designing advanced hybrid materials for next-generation supercapacitive energy storage.
1. Introduction
With the coming of energy crises and the increase of environmental problems, energy storage devices, especially supercapacitors, have received a great deal of attention because of their high power density, fast recharge capability, and long cycle life.1,2 Pseudocapacitors, as one of the two types of supercapacitor, have higher specific capacitance than the other (the EDLCs) owing to their fast faradaic reactions on the electrode surface.3,4Among various pseudocapacitive materials, nickel oxide (NiO) has attracted great interest due to its low cost, ease of synthesis, high theoretical specific capacitance of 2584 F g−1 and environmentally friendly.5 However, the intrinsic poor electrical conductivity and the low accessible surface areas of NiO severely limits its charge–discharge rate capability and power performance of electrode material for high-power applications.6,7
In order to solve this problem, various carbonaceous materials8–10 have been introduced to prepare the NiO-based composites and are expected to improve the electronic conductivity of electrodes. For example, Xia11 et al. reported a graphene sheet/porous NiO hybrid film by a combination of electrophoretic deposition and chemical-bath deposition, which showed a specific capacitance of 400 F g−1 at a current density of 2 A g−1. Bu12 et al. fabricated a NiO/rGO composite by a chemical reduction method, which displayed a specific capacitance of 461 F g−1 at a current density of 0.21 A g−1. Su13 et al. prepared a petal-like GNS/NiO composite via a microwave-assisted method, which exhibited a specific capacitance of 799 F g−1 at a current density of 0.3 A g−1. Jiang14 et al. synthesized a porous graphene/NiO composite by a hydrothermal route, which delivered a specific capacitance of 429.7 F g−1 at a current density of 0.2 A g−1. In general, all of these graphene–NiO composites exhibited an improved specific capacitance. However, cycle stability of these graphene–NiO composites is needed to be further improved. Besides, these graphene–NiO composites were mostly prepared by a hydrothermal route, which may result in poor dispersibility and a certain degree of aggregation.
Recently, exfoliation of layered double hydroxides (LDHs) into single layer has provided a new type of nanosheet with ultimate two-dimension anisotropy and positive charge. More importantly, these nanosheets could be used to construct functional hybrid materials.15–17 Nickel hydroxide, with a layer structure similar to LDHs, is quite promising to be exfoliated and to form 2D nickel hydroxide nanosheets.18,19
Inspired by this interesting phenomenon, the heterogeneous assembly of the oppositely charged nanosheets at the molecular layer was used to design and prepare the NiO/reduced graphene oxide composites. In the composites, NiO nanosheets are uniformly distributed on both sides of RGO nanosheets, effectively reducing the π–π restacking of the RGO sheets. Meanwhile, the introduction of RGO in the composite significantly improves the electronic conductivity of NiO, consequently providing more available electrochemically active surface area for enhancing energy storage. When evaluated as the electrode material for supercapacitor, the NiO/RGO electrode exhibits a high specific capacitance of 782 F g−1 at a current density of 0.5 A g−1 and an excellent cycling stability with 94.1% of the initial capacitance over 3000 cycles at 2 A g−1. The assembled NiO/RGO//AC asymmetric supercapacitor exhibits a remarkable energy density of 32.5 W h kg−1 at a power density of 375 W kg−1 and a high cycling stability with 92.7% retention after 3000 cycles at at 2 A g−1. The high electrochemical performances demonstrated that the NiO/RGO composite can be a promising electrode material for supercapacitor applications. More importantly, exfoliation of the nitrate-intercalated Ni(OH)2 was achieved in deionized water without any strippers and toxic organic solvents. Furthermore, the entire process of the NiO/RGO composite preparation has no rubbish and pollution, and the reaction condition is mild. Such a simple and green exfoliation–restacking method following with thermal treatment is promising to be applied to the preparation of other transition-metal oxide/RGO composites.
2. Experimental
2.1 Synthesis of graphene oxide nanosheets
Graphene oxide (GO) was prepared from natural graphite powders by a modified Hummer's method.20,21 The resultant material of graphite (1 g) after chemically oxidation and exfoliation was then dispersed in DI water to form a 0.5 mg mL−1 solution.2.2 Synthesis and exfoliation of Ni(OH)2
Ni(NO3)2·6H2O (2 g) was dissolved in DI water (200 mL). The solution pH value was adjusted to 9.0 by adding 25% NH3·H2O solution. After reaction for 6 h, the precipitate was achieved by vacuum filtration, washed with DI water and ethanol several times and redispersed in DI water. Exfoliation of the Ni(OH)2 was obtained by mechanical stirring then followed by ultrasonication (bath sonicator, 50 Hz, 1 h).2.3 Synthesis of the NiO/RGO composite
The dispersed Ni(OH)2 colloidal suspension was mixed with the GO solution, and the mixture was then stirred for 2 h. The brown precipitate containing the Ni(OH)2/GO composite was obtained by vacuum filtration and washed several times with DI water and dried by vacuum freeze-drying. Finally, as-prepared Ni(OH)2/GO composite was annealed at 300 °C for 4 h under an atmosphere of argon in order to obtain a NiO/RGO composite. A series of Ni(OH)2/GO composites with different GO mass fraction of 1%, 3% and 5% have been synthesized in above method, which were denoted as GON1, GON3 and GON5, respectively. Accordingly, NiO/RGO composites were denoted as GN1, GN3 and GN5, respectively. For comparison, pure NiO and RGO were prepared by the same process.2.4 Characterization
Phase structures of the products were characterized by X-ray diffraction (XRD, Bruker D8 Advance) with Cu Kα radiation (λ = 0.15406 nm). The morphology and microstructure of the products were examined by scanning electron microscopy (SEM, JSM-7001F) and transmission electron microscope (TEM, JEM-2010). The elemental mapping analyses of the composite were performed with an energy dispersive X-ray spectrometer equipped in the SEM machine. X-ray photoelectron spectroscopy (XPS) measurements were performed using an ESCALAB 250XI XPS with an Al Kα X-ray source (1486.6 eV). The specific surface area and the pore size distribution were evaluated from a N2 adsorption–desorption analysis conducted at 77 K on JW-BK122W. The specific surface area was estimated by the Brunauer–Emmett–Teller (BET) method, and pore size distribution was achieved from desorption plots by a Barrett–Joyner–Halenda (BJH) analysis. Zeta potential of the colloidal suspension was determined by the Malvern Zetasizer nano (ZS90, UK). Thermogravimetric analysis (TGA) of samples were performed on a NETZSCH STA409PC instrument, under air flow with a heating rate of 10 °C min−1, from room temperature to 800 °C.2.5 Electrochemical tests
The electrochemical measurements of the as-prepared electrodes were carried out by electrochemical workstation (CHI660E, Shanghai) in a three-electrode cell configuration and with a 6 M KOH as electrolyte. The three-electrode cell consisted of Pt foil as the counter electrode, Hg/HgO as the reference electrode. The working electrode was prepared by mixing the active material, acetylene black, and polyfluortetraethylene (PTFE) in a weight ratio of 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:5 and coated onto Ni foam (1.0 cm × 1.0 cm), and then dried in vacuum at 50 °C for 24 h. The average weight of the mixed materials was about 5 mg in each electrode.
:10:5 and coated onto Ni foam (1.0 cm × 1.0 cm), and then dried in vacuum at 50 °C for 24 h. The average weight of the mixed materials was about 5 mg in each electrode.
Cyclic voltammetry (CV) measurements were measured at various scanning rates between 0 and 0.5 V. Electrochemical impedance spectra (EIS) were performed at the amplitude of 5 mV in a frequency range from 100 kHz to 0.01 Hz. The galvanostatic charge and discharge measurements were carried out at the voltage range of 0–0.5 V.
The specific capacitance (Cs) is quantified from the galvanostatic discharge curves using the following eqn (1):22
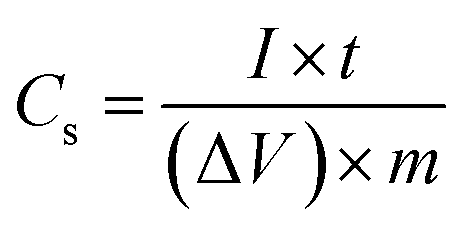 | (1) |
The asymmetric supercapacitor was investigated in a two-electrode system in 6 M KOH aqueous solution with GN3 as cathode and activated carbon as anode. The polypropylene membrane was utilized as separator. Activated carbon powder with a specific area of 1893 m2 g−1 was prepared from petroleum coke (Guangzhou, China) via reactions with KOH (mpetroleum coke:mKOH = 1:6) for 1.5 h at 850 °C. The proper mass ratio of active materials (GN3:AC) was calculated to be 1:1.53 based on their single-electrode capacitances. All the electrochemical measurements were measured on a CHI660E electrochemical workstation.
3. Results and discussion
Schematic illustration for the synthesis of NiO/RGO composite and the corresponding digital photographs are shown in Fig. 1. Firstly, the nitrate-intercalated Ni(OH)2 was prepared by a facile chemical precipitation method. Then, owing to a weak affinity of NO3− towards the Ni(OH)2 nanosheet,18 positively charged 2D Ni(OH)2 nanosheets were easily obtained in water with the assistance of ultrasonication. Exfoliation of the Ni(OH)2 sample yielded a light green and translucent colloidal solution (see Fig. 1b(1) and c). A clear Tyndall light scattering in Fig. 1c further indicated the presence of exfoliated Ni(OH)2 nanosheets in water. The effect of different anions on the exfoliation and stability of Ni(OH)2 were investigated. Fig. S1 and Table S1† indicated that NO3− ion is more favourable for the exfoliation and stability of Ni(OH)2, so this paper takes nickel nitrate as the source of nickel to prepare Ni(OH)2. GO nanosheets are highly negatively charged when dispersed in water, apparently as a result of ionization of the carboxyl, hydroxyl, and epoxyl groups bonded to carbon on the surface.23 A clear Tyndall light scattering in Fig. 1d showed the uniform dispersion of exfoliated GO nanosheets in water. The zeta potentials of two suspensions were +31.8 mV for Ni(OH)2 and −46 mV for GO respectively, indicating their good stability and excellent dispersion. When the two components with oppositely charge were mixed together and under continuous stirring, mutual electrostatic interactions drove the two substances to assemble into Ni(OH)2/GO precursor (see Fig. 1b(3)). Finally, the freeze-dried Ni(OH)2/GO precursor was heated at 300 °C for 4 h under an atmosphere of argon to obtain the NiO/RGO composite. During the thermal treatment, GO was reduced into reduced graphene oxide, while Ni(OH)2 in the assemblies was simultaneously converted into NiO. The final NiO/RGO composite shows layered structure. NiO nanosheets are uniformly and parallel distributed on both sides of RGO nanosheets, which effectively prohibited the restacking of individual RGO nanosheets, enhanced the conductivity of NiO, and formed a well-dispersed NiO/RGO composite. The synergistic effect between RGO and NiO resulted in rich porosity and large specific surface area for capacitor electrodes. A series of NiO/RGO composites with different mass ratio of NiO to RGO (GN1, GN3 and GN5) have been synthesized. The mass ration of NiO:RGO in GN1, GN3 and GN5 based on TGA curves (Fig. S3†) are about 92.9:7.1, 91.0:9.0 and 89.8:10.2, respectively. The electrochemical test of three NiO/RGO composites were shown in Fig. S2.† Obviously, GN3 exhibits the maximum specific capacitance and the most excellent electrochemical performance among the three NiO/RGO composites, so this paper use GN3 as representation to study the physicochemical properties of NiO/RGO composites.
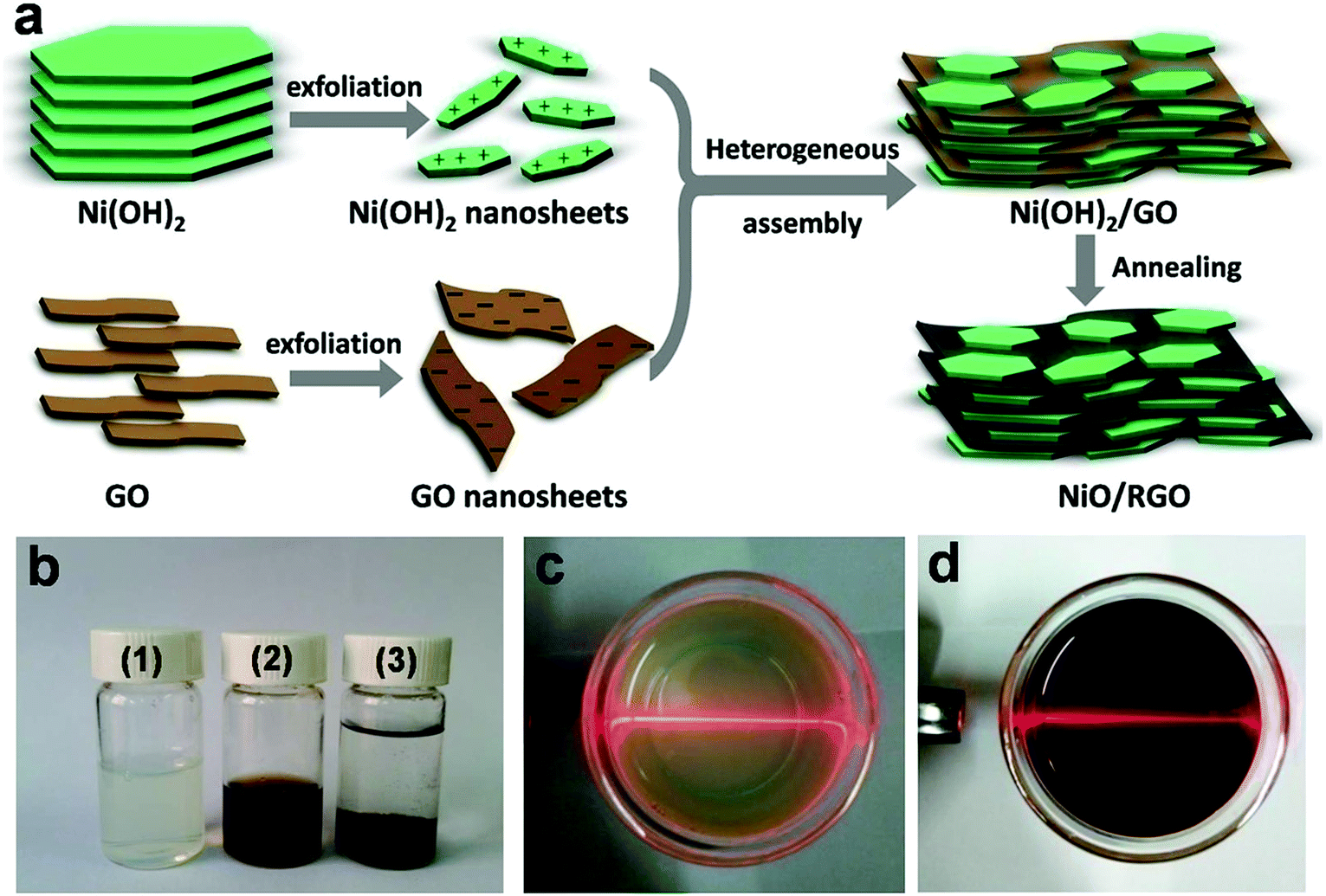 | ||
| Fig. 1 (a) Schematic illustration for the synthesis of NiO/RGO composite. (b) Digital images of the (1) exfoliated Ni(OH)2 nanosheets suspension, (2) GO nanosheets suspension and (3) Ni(OH)2/GO precursor. Tyndall light scattering of (c) the Ni(OH)2 nanosheets suspension and (d) GO nanosheets suspension. The light beam demonstrated the Tyndall effect. | ||
Fig. 2 shows the XRD patterns of freeze-drying GO, RGO, GN3 and pure NiO, respectively. Freeze-drying GO appeared two peaks at around 10.41° and 42.21°, which correspond to the (001) and (111) diffraction planes,24 and its interlayer spacing obtained from the Bragg formula is 0.88 nm. After reduction at 300 °C in argon for 4 h, the peak at 10.41° corresponding to GO completely disappeared, but a weak peak, broad peak (002) centered at around 24.5° was observed, which can be indexed into the disorderedly stacked RGO sheets.24 As seen in the XRD patterns of GN3 and pure NiO, the diffraction peaks corresponding to cubic NiO (JCPDS no. 47-1049) appearing at 37.2°, 43.2°, 62.8°, 75.2°, 79.4° can be indexed as (1 1 1), (2 0 0), (2 2 0), (3 1 1) and (2 2 2) crystal planes, respectively. In addition, the restacking diffraction peak of RGO nanosheets in GN3 at 24.5° is almost disappeared, indicating that NiO nanosheets uniformly distributed on both sides of the graphene sheets resulting in the low degree of graphitization.25
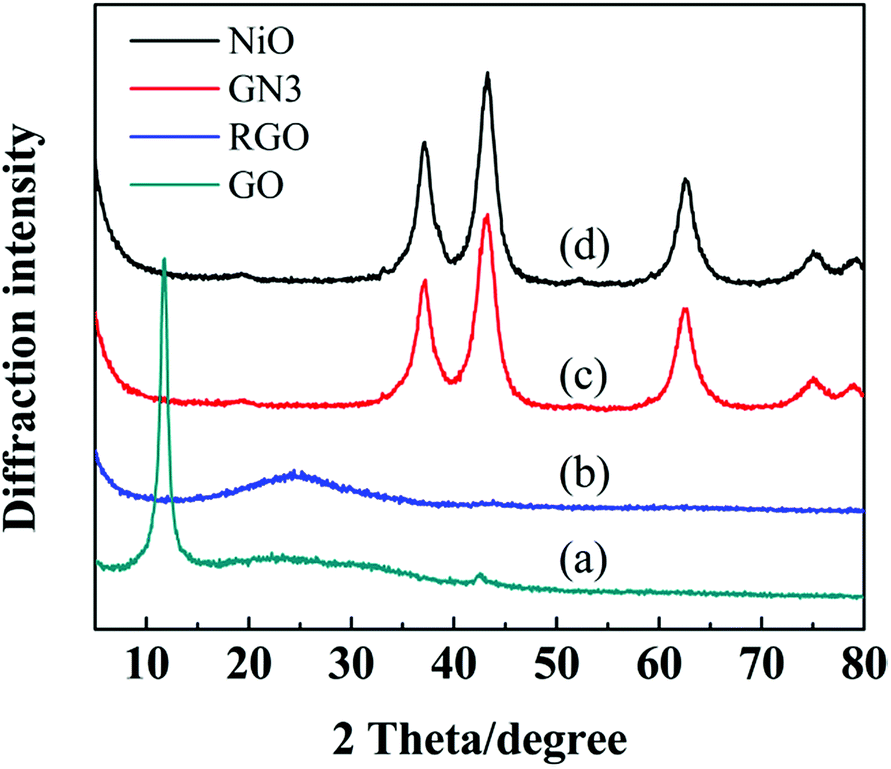 | ||
| Fig. 2 XRD patterns of (a) freeze-drying GO, (b) RGO, (c) GN3 and (d) pure NiO. | ||
The surface chemical compositions of the samples were investigated by XPS. Fig. 3a shows the XPS of GN3 with the binding energy ranges from 0 to 1200 eV. The peaks at 854.1 and 873 eV correspond to Ni 2p3/2 and Ni 2p1/2 spin-orbits,26 respectively. Fig. 3b compares the C 1s XPS of GO and GN3. The C 1s XPS of GO can be fitted into four peaks, corresponding to the four functional groups: carbon sp2 (C–C or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, 284.8 eV), carbon in C–O bonds (286.7 eV), carbonyl carbon (CO, 287.5 eV) and carboxylate carbon (O–CO, 290.3 eV).27 After the thermal treatment, the C–O, CO and O–CO groups in GO become very weak in the C 1s XPS spectra of GN3, indicating most oxygen containing groups have been successfully removed. The sufficient reduction was also confirmed by the result of XRD analysis shown in Fig. 2. Fig. 3c shows the O 1s spectra of GN3, which can be divided into two peaks. The peak at 529.3 eV is corresponding to the oxygen bonded with nickel in NiO, and the peak at 531.2 eV is assigned to the residual oxygen-containing group in RGO.28 The Ni 2p3/2 and Ni 2p1/2 peaks are showed in Fig. 3d: 2p3/2 (854.1 eV) and its satellites (855.8 and 861 eV), 2p1/2 (873 eV) and its satellite (879.5 eV), all of which can be assigned to the Ni phase of NiO. These results are also consistent with the XRD.
C, 284.8 eV), carbon in C–O bonds (286.7 eV), carbonyl carbon (CO, 287.5 eV) and carboxylate carbon (O–CO, 290.3 eV).27 After the thermal treatment, the C–O, CO and O–CO groups in GO become very weak in the C 1s XPS spectra of GN3, indicating most oxygen containing groups have been successfully removed. The sufficient reduction was also confirmed by the result of XRD analysis shown in Fig. 2. Fig. 3c shows the O 1s spectra of GN3, which can be divided into two peaks. The peak at 529.3 eV is corresponding to the oxygen bonded with nickel in NiO, and the peak at 531.2 eV is assigned to the residual oxygen-containing group in RGO.28 The Ni 2p3/2 and Ni 2p1/2 peaks are showed in Fig. 3d: 2p3/2 (854.1 eV) and its satellites (855.8 and 861 eV), 2p1/2 (873 eV) and its satellite (879.5 eV), all of which can be assigned to the Ni phase of NiO. These results are also consistent with the XRD.
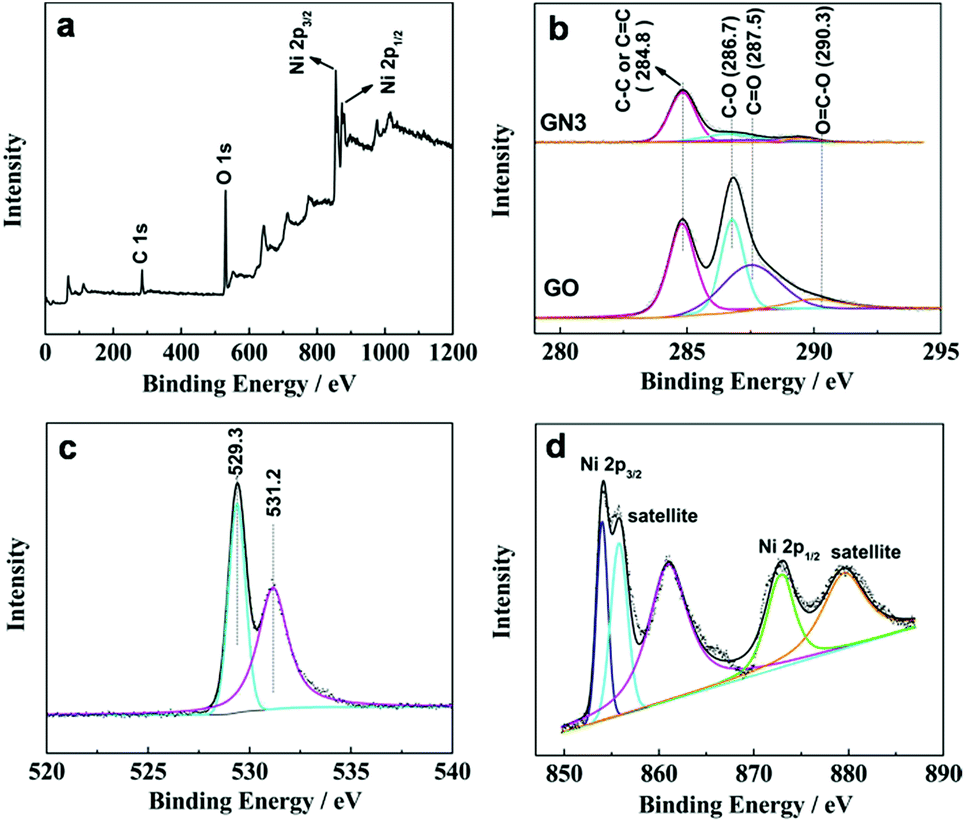 | ||
| Fig. 3 (a) XPS of GN3, (b) C 1s XPS of GO and GN3, (c) O 1s XPS of GN3 and (d) Ni 2p XPS of GN3. | ||
The morphology and microstructure of the as-obtained products are shown in Fig. 4. It can be seen from Fig. 4a that the RGO is crumpled to a curly and wavy shape, and few layers of the graphene are stacked with each other through van der Waals interactions of the remaining oxygen-containing functionalities on the surfaces of the graphene.29,30 The SEM image of pure NiO (Fig. 4b) shows that the NiO is consisted of irregular layers and these layers arrange randomly to form the agglomerates. To show the heterogeneous assembly and the coexistence of GO and Ni(OH)2, a cross sectional SEM image of GON3 was obtained as shown in Fig. 4c. It is clear that the Ni(OH)2 nanosheets are restacking on the surface of the GO driven by the electrostatic interaction, and the introduction of GO effectively prevents the aggregation of layered Ni(OH)2, which confirms our presumed formation mechanism. Fig. 4d displays the cross-sectional SEM image of GN3 obtained by the thermal treatment of GON3. During the whole annealing process, it could be seen that the multi-layered structure with most of the sheets parallel to each other was approximately maintained, and the composite is loose and not compactly packed. Such a structure guarantees that the electrolyte ions can efficiently diffuse into the loose interlayer regions and further contact with the composite surface area effectively, and consequently the composite provides more redox faradaic reactions for enhancing energy storage.
 | ||
| Fig. 4 SEM images of (a) RGO, (b) pure NiO, (c) GON3 and (d) GN3. | ||
A cross sectional SEM image of the GN3 and the corresponding elemental mapping images of carbon, oxygen, and nickel were shown in Fig. 5. All of the carbon (Fig. 5b), oxygen (Fig. 5c) and nickel (Fig. 5d) elements are homogeneously distributed in the as-prepared GN3 composite, indicating the heterogeneous assembly and coexistence of NiO and RGO in the product, which was identical with the XRD results. This kind of configuration make the GN3 has better electrochemical performance than pure NiO, which will be shown in the later parts.
 | ||
| Fig. 5 Cross sectional SEM image of (a) GN3 and the corresponding elemental mapping images of (b) carbon, (c) oxygen, and (d) nickel. | ||
To further characterize the structure of the GN3 composite, TEM studies were carried out (Fig. 6). From TEM images in Fig. 6a, the exfoliated Ni(OH)2 in the colloidal suspensions exhibits irregular and thin hexagonal platelets with a mean lateral size of 80 nm. TEM image of the exfoliated GO in Fig. 6b shows some wrinkles and folds, which result from the fact that the 2D membrane structure becomes thermodynamically stable via bending. The image of the Ni(OH)2/GO composite (GON3) is shown in Fig. 6c. In comparison with the TEM image of GO, the surface of the composite appears many small irregular nanosheets, which can be attributed to the anchoring of Ni(OH)2 nanosheets on GO. After annealing treatment, it could be seen in Fig. 6d that the NiO/RGO composite (GN3) almost retained the original morphology of its precursor Ni(OH)2/GO composite, which is consistent with the result of SEM. The final NiO/GO composite displays that the NiO nanosheets are uniformly dispersed on the surface of RGO.
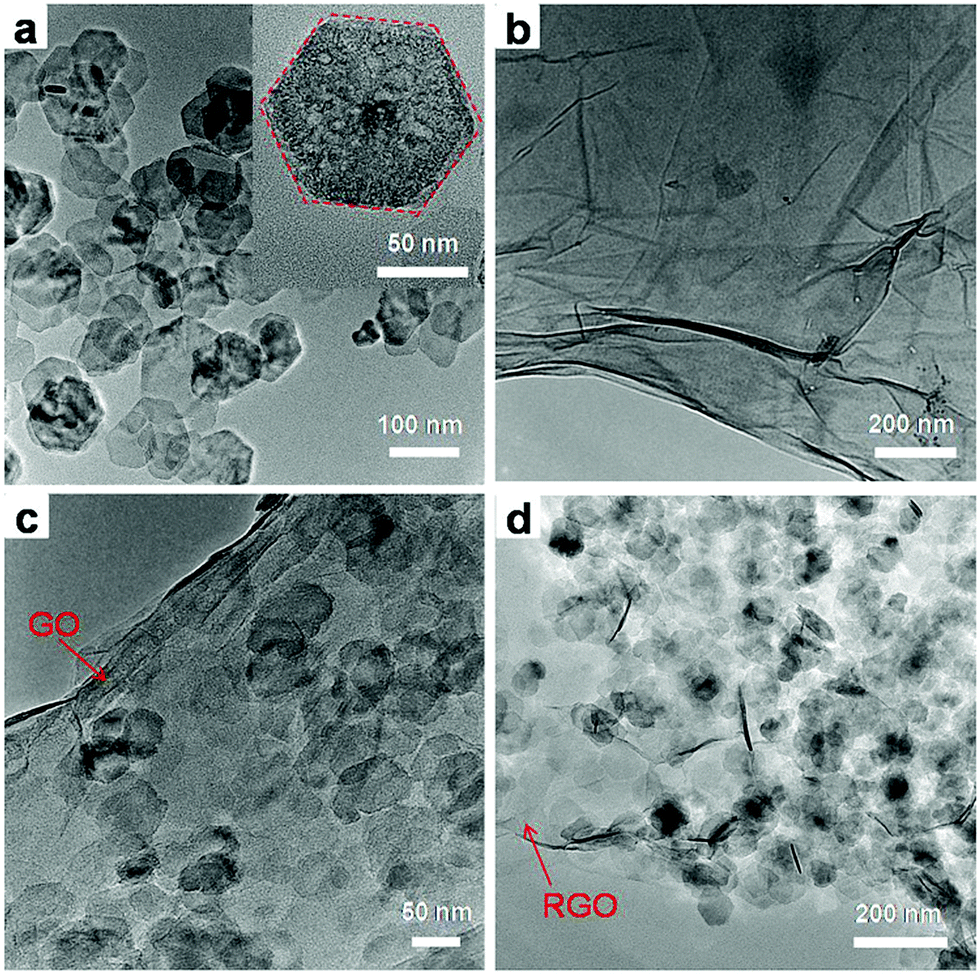 | ||
| Fig. 6 TEM images of (a) exfoliated Ni(OH)2 nanosheets, and (b) exfoliated GO, (c) GON3 and (d) GN3. | ||
The nitrogen adsorption/desorption isotherms and the pore size distribution plots of pure NiO and GN3 are shown in Fig. 7. According to IUPAC classification, GN3 displayed a type IV isotherm loop. The specific surface area of pure NiO and GN3 were measured by nitrogen gas adsorption. The specific surface area (SSA) of GN3 is calculated to be 143.1 m2 g−1, which is larger than that of pure NiO (65.3 m2 g−1). This is mostly because of the heterogeneous assembly of RGO and NiO, and thus NiO nanosheets can be separated one by one. Therefore, NiO nanosheets on both sides of RGO nanosheets become smaller than pure NiO, and the specific surface area is expected to become larger.31,32 Besides, the RGO also contribute to the specific surface area to a certain extent. Fig. 7b shows the pore distribution of NiO and GN3. The pore diameter of NiO is mainly appeared at around 20 nm, while the pores of GN3 are mainly around 2–5 nm. Heterogeneous assembly of RGO and NiO result in loose multi-layered structure of NiO/RGO composite. RGO nanosheets partially overlap with each other to form a three-dimensional conductive network, and the NiO nanosheets are uniformly distributed on both sides of RGO nanosheets, thus the NiO/RGO composite shows homogeneous pore size distribution. The three-dimensional conductive RGO network, homogeneous pore size distribution and the large specific surface area provide more tunnels for electron and ion transport to enhance the electrochemical performance of the GN3 electrode.
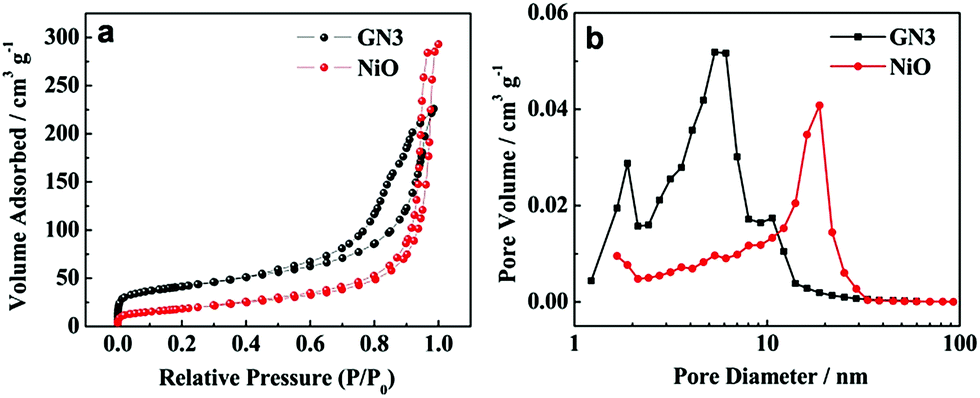 | ||
| Fig. 7 (a) Nitrogen adsorption/desorption isotherms of pure NiO and GN3, (b) pore size distribution patterns of pure NiO and GN3. | ||
The electrochemical properties of the NiO/RGO composite are measured by cyclic voltammogram (CV) and galvanostatic discharge in a three-electrode cell configuration. Fig. 8a shows the CV curves of RGO, NiO, GN3 samples at a scanning rate of 20 mV s−1 in 6 M KOH solution. A pair of redox peaks can be observed in the CV curves of NiO and GN3, which is mainly derived from the surface faradaic reaction of NiO as shown in the following formula:33
| NiO + OH− ↔ NiOOH + e− | (2) |
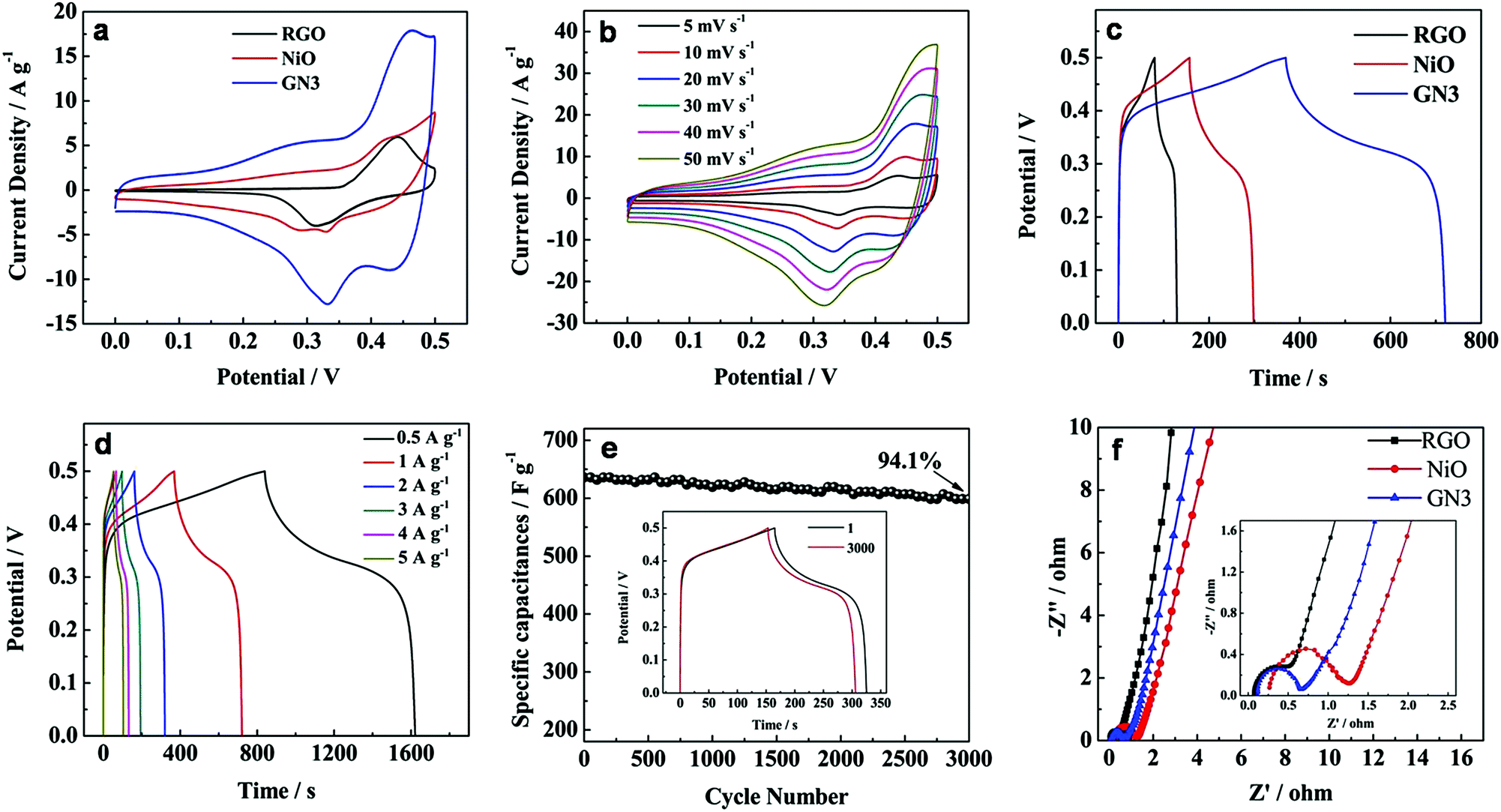 | ||
| Fig. 8 (a) CV curves of RGO, NiO, GN3 samples at a scanning rate of 20 mV s−1 in 6 M KOH solution, (b) CV curves of GN3 at different scan rates in 6 M KOH solution, (c) the galvanostatic charge–discharge curves of RGO, NiO, GN3 at the current density of 1 A g−1, (d) the galvanostatic charge–discharge curves of GN3 at different discharge current densities, (e) cycling performance of GN3 at a current density of 2 A g−1 in a 6 M KOH solution. The inset shows the 1st and 3000th charge–discharge curves of GN3 at a current density of 2 A g−1, (f) Nyquist plots of GN3, RGO, NiO electrodes in the frequency range of 100 kHz to 0.01 Hz. The inset shows the enlarged EIS of the electrodes. | ||
The CV curve of GN3 is larger than that of pure NiO at the same scan rate, indicating the larger specific capacitances. It can be interpreted that the introduction of RGO in the composite effectively improved the conductivity of the NiO, and consequently provided more available active sites for redox reactions and thus the pseudocapacitive behavior of NiO could be fully fulfilled. Furthermore, it is noted that a pair of redox peaks also can be observed from the CV curve of RGO, which is ascribed to the redox reaction of OH− with the residual functional groups in RGO.34 Therefore, RGO in the GN3 composite can make extra contributions to the enhanced electrochemical performance of GN3 composite. Each CV curve of GN3 (Fig. 8b) at different scan rates in 6 M KOH solution maintains similar redox couples. As the scan rates increase, the shape of CV curves has little change and polarization, indicating GN3 maintains good rate capability. Besides, it can be seen that the oxidation and reduction peaks shifted positively and negatively respectively with the scan rate increased because of the internal resistance.35
Fig. 8c shows the charge–discharge curves of RGO, NiO, GN3 at the current density of 1 A g−1. The specific capacitance of GN3 at 1 A g−1 was calculated to be 704 F g−1, which is much larger than that of RGO (98 F g−1) an NiO (282 F g−1). Given the excellent rate capability of the GN3 sample, we specially investigated its charge–discharge curves at the current density of 0.5, 1, 2, 3, 4 and 5 A g−1 (Fig. 8d). The specific capacitance of GN3 calculated from the discharge curves (Fig. 8d) was 782, 704, 636, 576, 528 and 510 F g−1, respectively. Furthermore, the excellent cycle stability of the NiO/RGO composite electrode is also demonstrated in Fig. 8e. Over 3000 cycles, the specific capacitance of the GN3 composite slightly decreased from 636 to 599, with a 94.1% capacitance retention ratio of the initial capacity (the inset in Fig. 8e). These results reveal that the high specific capacitance, remarkable rate capability and excellent cycle stability are achieved in the NiO/RGO composite for electrochemical supercapacitors.
Electrochemical impedance spectroscopy (EIS) is also an significant chemical analytic technique to examine the fundamental behavior of electrode materials for supercapacitors. Fig. 8f shows the Nyquist plots of GN3, RGO and NiO electrodes with a frequency range from 100 kHz to 0.01 Hz at an amplitude of 5 mV. Each plot was characterized by a depressed semicircle in the high frequency region and an inclined line in the low frequency region.36,37 The intercept on the real axis in the high frequency range is accepted to the bulk solution resistance (Re). The semicircle corresponds to the charge-transfer resistance (Rct) at the electrode/electrolyte interface, and the numerical value of the diameter of the semicircle on the real axis is approximately equal to Rct. The inclined line in the low frequency region corresponds to the ion diffusion process within the electrodes. The calculated values of Re are 0.07, 0.26 and 0.12 Ω for RGO, NiO and GN3, respectively. It is clear that the Re of GN3 is a much lower than that of NiO due to the introduction of RGO. It is well accepted that a larger semicircle means a larger charge transfer resistance, and a steeper slope signifies a faster ion-diffusion rate. Obviously, the semicircle of RGO is the smallest, showing that Rct of RGO is significantly low due to the high conductivity of RGO. The calculated values of Rct are 1.04 and 0.56 Ω for NiO and GN3, respectively. Compared with the pure NiO electrode, the GN3 composite electrode exhibited a smaller semicircle and a steeper slope, suggesting that the GN3 composite electrode had a lower Rct and faster ion diffusion rate than the pure NiO electrode.
The excellent specific capacitance and cycling performance can be attributed to the heterogeneous assembly and the coexistence of RGO and NiO. First, the controlled heteroassembly of the oppositely charged nanosheets and the subsequent in situ thermal reduction are favorable for the formation of graphene-based hybrid materials with nanoscale dispersion and high interfacial interaction between the NiO matrix and graphene sheets. Second, heterogeneous assembly of RGO and NiO result in loose multi-layered structure of NiO/RGO composite, which creates abundant space, further decreasing the kinetic limitations of pseudo-capacitive electrode, and hence resulting in the high energy storage. Third, the presence of RGO in the layered structure prevents the aggregation of NiO nanosheets and simultaneously enhances the conductivity of the composite, which provides more available active sites for redox reactions and thus the pseudocapacitive behavior of NiO in the composite can be better displayed. The loose multi-layered structure and the coexistence of RGO and NiO both enhance the capacity and cycling performance of the GN3 electrode. As a result, the NiO/RGO composite shows good rate capabilities and excellent cycling performance.
To further investigate the practical application of the GN3 composite material, an asymmetric supercapacitor cell was assembled by utilizing the GN3 as cathode and activated carbon (AC) as anode with a polypropylene separator (denoted as GN3//AC) (Fig. 9a). To determine the stable electrochemical windows of the asymmetric supercapacitor device, the CV curves of GN3 and AC electrodes were measured in 6 M KOH solution, respectively, as shown in Fig. 9b. According to the CV curves, the GN3 electrode was measured within a stable potential window of 0–0.5 V, while a stable potential window of the AC electrode was measured from −1.0 to 0 V, which indicates that the GN3//AC asymmetric supercapacitor device can achieve a maximum working voltage of 1.5 V in 6 M KOH. The pore structure and the electrochemical test of AC were shown in Fig. S4 and S5,† respectively.
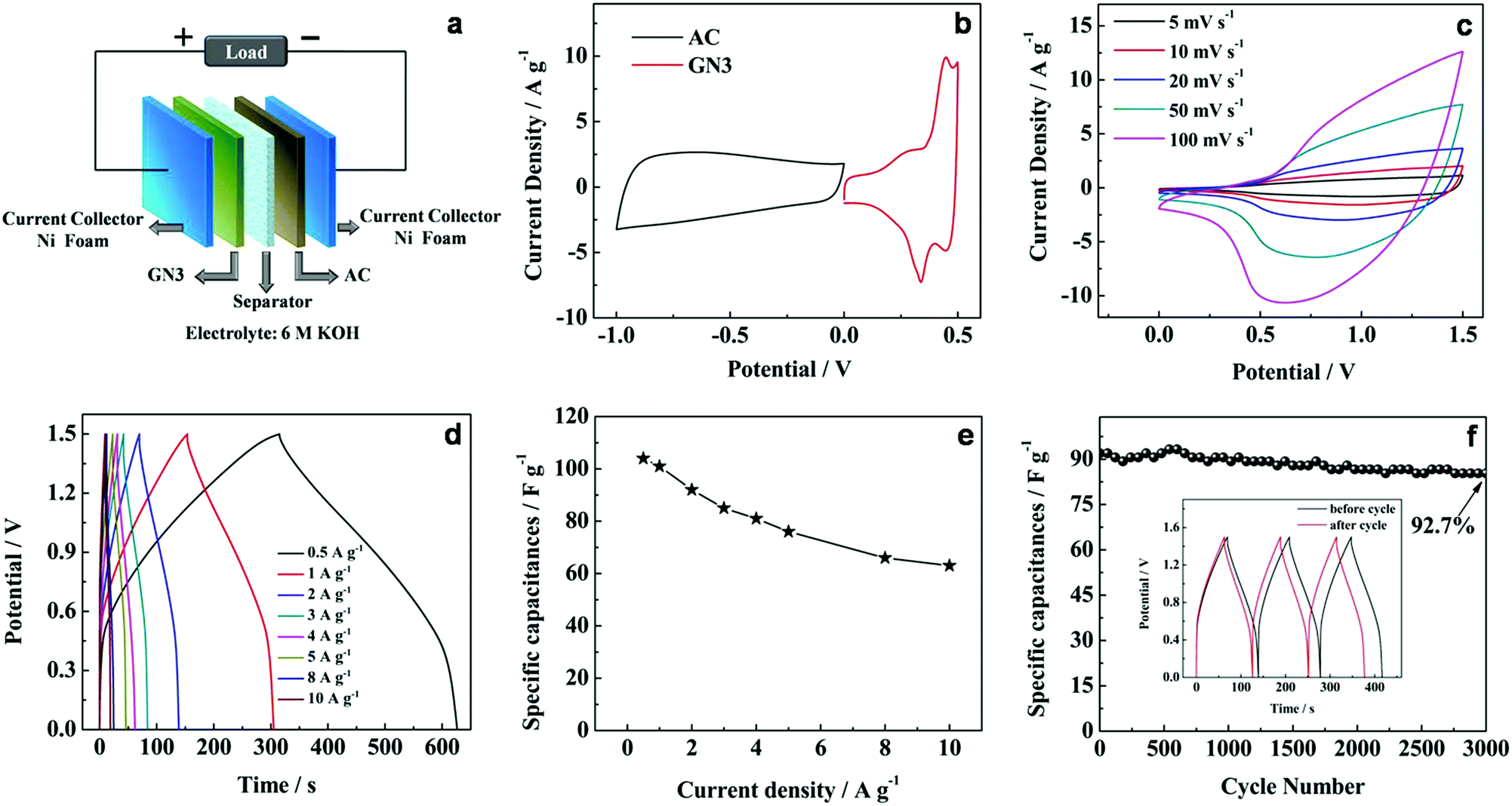 | ||
| Fig. 9 (a) Schematic illustration of the assembled structure of the GN3//AC asymmetric supercapacitor, (b) CV curves of GN3 and AC electrodes at a scan rate of 10 mV s−1 in 6 M KOH solution. Electrochemical characterization of the GN3//AC asymmetric supercapacitor in 6 M KOH solution, (c) CV curves at different scan rates, (d) galvanostatic charge–discharge curves at different current densities, (e) corresponding specific capacitance at different current densities, (f) cycling performance at 2 A g−1 for 3000 cycles. The inset shows the corresponding galvanostatic charge–discharge curves of the initial 3 cycles and the last 3 cycles. | ||
In the asymmetric supercapacitor cell, the charge balance should follow the equation:35,38
| Q+ = Q− | (3) |
The stored charge (Q) by each electrode is related to the specific capacitance (C), the potential window in the three-electrode measurement (ΔV), and the weight of each electrode (m), following the relationship:
| Q = C × ΔV × m | (4) |
In order to get the charge balance, the mass loading ratio of cathode and anode can be estimated by the following equation:
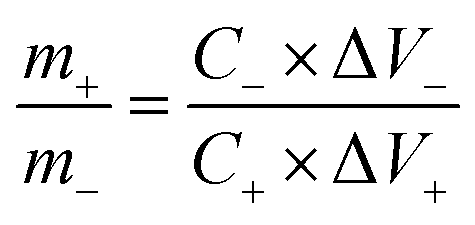 | (5) |
In our case, the specific capacitances calculated from the galvanostatic charge–discharge curves are 704 F g−1 and 230 F g−1 for GN3 and AC at 1 A g−1, respectively. So the mass loading ratio of the cathode (GN3, 3 mg) to anode (AC, 4.59 mg) was adjusted to be 1:1.53.
Fig. 9c shows the CV curves of the GN3//AC asymmetric supercapacitor at different scan rates between 0 and 1.5 V in 6 M KOH solution. The broad redox peaks were observed, which indicates that the faradaic pseudocapacitive behavior of the GN3//AC asymmetric capacitor induced by the GN3 electrode. In addition, the shape of the CV curves can still be maintained with increasing scan rates, and no obvious distortion was observed even at a high scan rate of 100 mV s −1, indicating the fast charge–discharge.
The galvanostatic charge–discharge plots at different current densities between 0 and 1.5 V are shown in Fig. 9d. No apparent IR drop is observed for all the curves, indicating a low internal resistance for the asymmetric supercapacitor. Moreover, all the discharge curves are nearly symmetric to their corresponding charging counterparts, which suggested that the as-assembled cell has excellent electrochemical reversibility and capacitive characteristics. The specific capacitance was calculated based on the total mass loading of the active materials on both electrodes (Fig. 9e and Table S2†). The specific capacitance achieves 104 F g−1 at 0.5 A g−1, and retains 63.3 F g−1 at 10 A g−1, implying good rate performance.
Additionally, the GN3//AC asymmetric supercapacitor exhibited an good cycling stability with 92.7% specific capacitance retention after 3000 cycles at 2 A g−1 (Fig. 9f). The cycling performance outperforms many other previously reported samples, such as porous NiO//carbon (50% retention after 1000 cycles),39 NiCo2O4/reduced graphite oxide//activated carbon (83% retention after 2500 cycles)40 and NiO//RGO (88% retention after 2000 cycles).41 After cycling, the last 3 cycles are almost symmetric and nearly identical to the initial 3 cycles (inset in Fig. 9f). The excellent capacitive performance of the GN3//AC asymmetric supercapacitor could be related to the charge balance and synergistic effect between cathode and anode.
Fig. 10 shows a Ragone plot to estimate the energy and power property of the GN3//AC asymmetric supercapacitor.
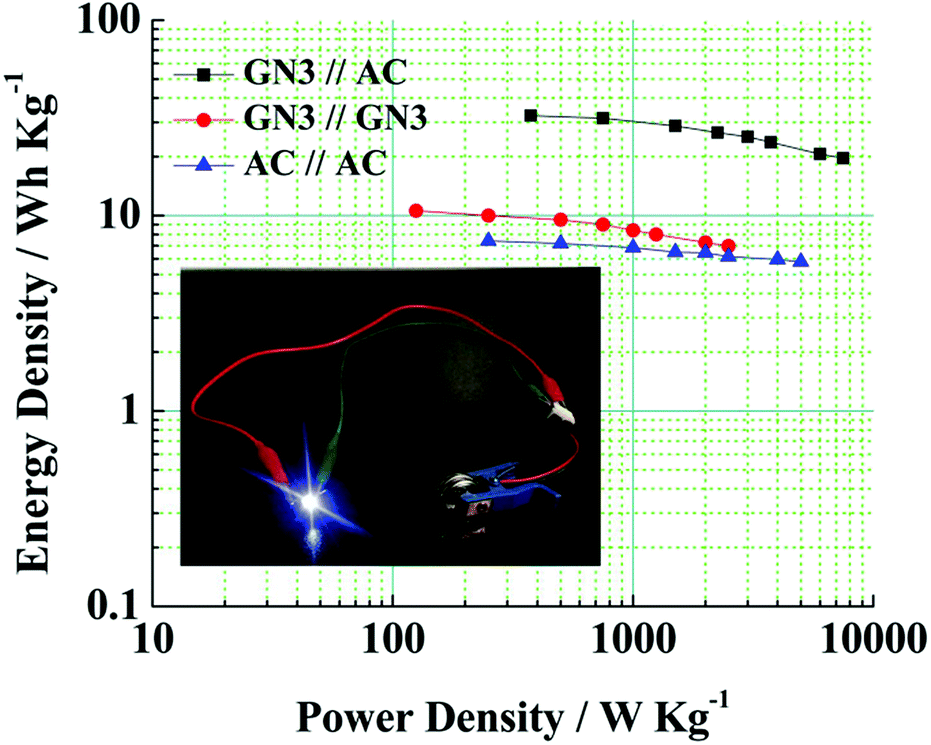 | ||
| Fig. 10 Ragone plots of the GN3//AC asymmetric supercapacitor, GN3//GN3 symmetric supercapacitor and AC//AC symmetric supercapacitor. Inset shows a digital photograph of the lighted LED by two asymmetric supercapacitors in series. | ||
The energy density and power density of a asymmetric supercapacitor can be calculated from the following equations:
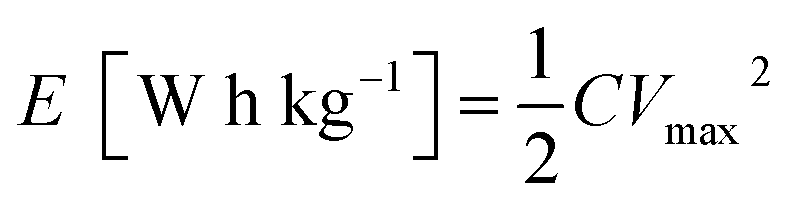 | (6) |
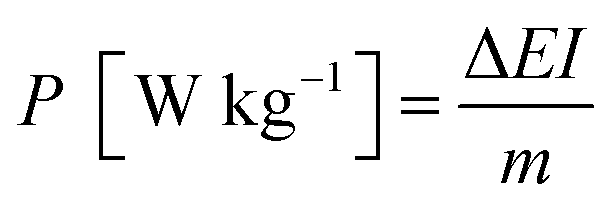 | (7) |
Obviously, the energy density of the GN3//AC asymmetric supercapacitor reaches 32.5 W h kg−1 at a power density of 375 W kg−1, and even retains 19.78 W h kg−1 at a power density of 7500 W kg−1 (Fig. 10 and Table S2†), which is much higher than those of the GN3//GN3 and the AC//AC symmetric supercapacitors. What's more, the energy and power densities of the GN3//AC asymmetric supercapacitor is better than or at least comparable to that of other reported symmetric supercapacitors and asymmetric supercapacitors, such as RuO2//RuO2 (15 W h kg−1 at 10 kW kg−1),42 Fe2O3/FGS//MnO2/FGS asymmetric (10 W h kg−1 at 32 kW kg−1),43 CNT@NiO//PCPs (25.4 W h kg−1 at 400 W kg−1),44 NiO//AC (14.6 W h kg−1 at 118 W kg−1)45 and NiO-dots/Gh//AC (27.3 W h kg−1 at 1562.6 W kg−1).46 The excellent electrochemical performance of the GN3//AC asymmetric supercapacitor can be due to the following aspects: (i) the loose multi-layered structure of GN3 composite prevents the aggregation of NiO nanosheets and the π–π restacking of RGO sheets, consequently resulting in a higher specific surface area, superior electrical conductivity and more available active sites, and then resulting in a large capacitance of anode. (ii) The AC with superior specific surface area, large pore volume and suitable pore size distribution as the anode is favorable for fast electrolyte ions transport, ensuring high power density and good rate capability. (iii) The GN3//AC asymmetric supercapacitor has a wide voltage window (1.5 V), consequently resulting in a great increase in energy density. Furthermore, two GN3//AC asymmetric supercapacitors connected in series can successfully light a LED (3.0 V), as shown in the inset of Fig. 10, implying its potential in electrochemical energy storage.
4. Conclusions
In summary, a NiO/RGO composite with well-controlled loose multi-layered structure has been successfully synthesized by a heterogeneous assembly approach with subsequent thermal treatment. In the hybrid architecture, graphene materials can be incorporated into hexagonal nanostructures to form high surface-area, conductive porous networks. In consideration of fast ionic and electronic transport, the NiO/RGO composite provides tremendous potential for energy storage application. As evaluated in a three electrode system, the optimal NiO/RGO electrode (GN3) exhibits a high specific capacitance (782 F g−1 at 0.5 A g−1) and an excellent cycling stability (94.1% retention after 3000 cycles at 2A g−1). A high performance GN3//AC asymmetric supercapacitor with the GN3 cathode and AC anode has been further demonstrated. The as-assembled GN3//AC asymmetric supercapacitor shows a remarkable energy density of 32.5 W h kg−1 at a power density of 375 W kg−1 and retained 19.78 W h kg−1 at a power density of 7500 W kg−1. Additionally, the asymmetric supercapacitor also exhibits an excellent cycling stability with 92.7% retention after 3000 cycles at 2 A g−1. This work indicates that the method we have developed is highly effective for the construction of advanced graphene based composites towards high-performance supercapacitor applications. More importantly, the facile, green and effective method described above should be readily applicable to the preparation of other transition-metal oxide/reduced graphene oxide composites.Acknowledgements
We gratefully acknowledge the support of this work by National Nature Science Foundation of China (No. 51402324, 51402325) and Natural Science Foundation of Shanxi Province (2015021077).References
- C. Liu, Z. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863–4868 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- W. Jiang, D. Yu, Q. Zhang, K. Goh, L. Wei, Y. Yong, R. Jiang, J. Wei and Y. Chen, Adv. Funct. Mater., 2015, 25, 1063–1073 CrossRef CAS.
- Z.-S. Wu, D.-W. Wang, W. Ren, J. Zhao, G. Zhou, F. Li and H.-M. Cheng, Adv. Funct. Mater., 2010, 20, 3595–3602 CrossRef CAS.
- G. Zhang, L. Yu, H. E. Hoster and X. W. Lou, Nanoscale, 2013, 5, 877–881 RSC.
- M. Zhi, C. Xiang, J. Li, M. Li and N. Wu, Nanoscale, 2013, 5, 72–88 RSC.
- G. Lee, Y. Cheng, C. V. Varanasi and J. Liu, J. Phys. Chem. C, 2014, 118, 2281–2286 CAS.
- Y. Bai, M. Du, J. Chang, J. Sun and L. Gao, J. Mater. Chem. A, 2014, 2, 3834–3840 CAS.
- B. Gao, C. Yuan, L. Su, S. Chen and X. Zhang, Electrochim. Acta, 2009, 54, 3561–3567 CrossRef CAS.
- C.-C. Lin and C.-C. Yen, J. Appl. Electrochem., 2008, 38, 1677–1681 CrossRef CAS.
- X. Xia, J. Tu, Y. Mai, R. Chen, X. Wang, C. Gu and X. Zhao, Chemistry, 2011, 17, 10898–10905 CrossRef CAS PubMed.
- Y. Bu, S. Wang, H. Jin, W. Zhang, J. Lin and J. Wang, J. Electrochem. Soc., 2012, 159, A990–A994 CrossRef CAS.
- X. Su, H. Chai, D. Jia, S. Bao, W. Zhou and M. Zhou, New J. Chem., 2013, 37, 439–443 RSC.
- Y. Jiang, D. Chen, J. Song, Z. Jiao, Q. Ma, H. Zhang, L. Cheng, B. Zhao and Y. Chu, Electrochim. Acta, 2013, 91, 173–178 CrossRef CAS.
- Z. P. Liu, R. Z. Ma, M. Osada, N. Iyi, Y. Ebina, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2006, 128, 4872–4880 CrossRef CAS PubMed.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Science, 2013, 340, 1226419 CrossRef.
- Q. Wu, A. O. Sjåstad, r. B. Vistad, K. D. Knudsen, J. Roots, J. S. Pedersen and P. Norby, J. Mater. Chem., 2007, 17, 965–971 RSC.
- S. Ida, D. Shiga, M. Koinuma and Y. Matsumoto, J. Am. Chem. Soc., 2008, 130, 14038–14039 CrossRef CAS PubMed.
- G. Lee, C. V. Varanasi and J. Liu, Nanoscale, 2015, 7, 3181–3188 RSC.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- Y. Wimalasiri and L. Zou, Carbon, 2013, 59, 464–471 CrossRef CAS.
- S.-J. Bao, C. M. Li, C.-X. Guo and Y. Qiao, J. Power Sources, 2008, 180, 676–681 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- D. Pan, S. Wang, B. Zhao, M. Wu, H. Zhang, Y. Wang and Z. Jiao, Chem. Mater., 2009, 21, 3136–3142 CrossRef CAS.
- D. Cai and M. Song, J. Mater. Chem., 2007, 17, 3678–3680 RSC.
- N. Spinner and W. E. Mustain, Electrochim. Acta, 2011, 56, 5656–5666 CrossRef CAS.
- H.-K. Jeong, Y. P. Lee, R. J. W. E. Lahaye, M.-H. Park, K. H. An, I. J. Kim, C.-W. Yang, C. Y. Park, R. S. Ruoff and Y. H. Lee, J. Am. Chem. Soc., 2008, 130, 1362–1366 CrossRef CAS PubMed.
- Z. S. Wu, W. C. Ren, L. Wen, L. B. Gao, J. P. Zhao, Z. P. Chen, G. M. Zhou, F. Li and H. M. Cheng, ACS Nano, 2010, 4, 3187–3194 CrossRef CAS PubMed.
- M. J. McAllister, J.-L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS.
- J.-L. Shi, G.-L. Tian, Q. Zhang, M.-Q. Zhao and F. Wei, Carbon, 2015, 93, 702–712 CrossRef CAS.
- H. Liu, G. X. Wang, J. Liu, S. Z. Qiao and H. J. Ahn, J. Mater. Chem., 2011, 21, 3046–3052 RSC.
- B. Zhao, J. Song, P. Liu, W. Xu, T. Fang, Z. Jiao, H. Zhang and Y. Jiang, J. Mater. Chem., 2011, 21, 18792–18798 RSC.
- J. A. Yan, E. Khoo, A. Sumboja and P. S. Lee, ACS Nano, 2010, 4, 4247–4255 CrossRef CAS PubMed.
- L.-J. Xie, J.-F. Wu, C.-M. Chen, C.-M. Zhang, L. Wan, J.-L. Wang, Q.-Q. Kong, C.-X. Lv, K.-X. Li and G.-H. Sun, J. Power Sources, 2013, 242, 148–156 CrossRef CAS.
- J. Yan, Z. Fan, W. Sun, G. Ning, T. Wei, Q. Zhang, R. Zhang, L. Zhi and F. Wei, Adv. Funct. Mater., 2012, 22, 2632–2641 CrossRef CAS.
- X. Zhao, Q. Zhang, C.-M. Chen, B. Zhang, S. Reiche, A. Wang, T. Zhang, R. Schlögl and D. Sheng Su, Nano Energy, 2012, 1, 624–630 CrossRef CAS.
- Y. Yao, C. Ma, J. Wang, W. Qiao, L. Ling and D. Long, ACS Appl. Mater. Interfaces, 2015, 7, 4817–4825 CAS.
- L. Kong, C. Zhang, S. Zhang, J. Wang, R. Cai, C. Lv, W. Qiao, L. Ling and D. Long, J. Mater. Chem. A, 2014, 2, 17962–17970 CAS.
- D.-W. Wang, F. Li and H.-M. Cheng, J. Power Sources, 2008, 185, 1563–1568 CrossRef CAS.
- X. Wang, W. S. Liu, X. Lu and P. S. Lee, J. Mater. Chem., 2012, 22, 23114–23119 RSC.
- X. Ren, C. Guo, L. Xu and T. Li, ACS Appl. Mater. Interfaces, 2015, 7, 19930–19940 CAS.
- H. Xia, Y. Shirley Meng, G. Yuan, C. Cui and L. Lu, Electrochem. Solid-State Lett., 2012, 15, A60 CrossRef CAS.
- H. Xia, C. Hong, B. Li, B. Zhao, Z. Lin, M. Zheng, S. V. Savilov and S. M. Aldoshin, Adv. Funct. Mater., 2015, 25, 627–635 CrossRef CAS.
- H. Yi, H. Wang, Y. Jing, T. Peng and X. Wang, J. Power Sources, 2015, 285, 281–290 CrossRef CAS.
- G. Cheng, Q. Bai, C. Si, W. Yang, C. Dong, H. Wang, Y. Gao and Z. Zhang, RSC Adv., 2015, 5, 15042–15051 RSC.
- M. Jing, C. Wang, H. Hou, Z. Wu, Y. Zhu, Y. Yang, X. Jia, Y. Zhang and X. Ji, J. Power Sources, 2015, 298, 241–248 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: SEM and TEM images, BET, XPS spectra, XRD patterns, electrochemical capacitive performances, and tables. See DOI: 10.1039/c6ra04998b |
| ‡ Both authors are co-first authors. |
| This journal is © The Royal Society of Chemistry 2016 |