
Impact of chelation on anticancer activities of organometallic ruthenium(ii) complexes containing 2,5-di(1H-pyrazol-1-yl)-1,4-benzoquinone: synthesis, structure, DNA/protein binding, antioxidant activity and cytotoxicity†
Abstract
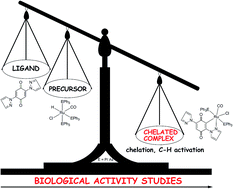
To gain some insight into the influence of chelation on anticancer activities, two new bivalent organometallic ruthenium complexes, [Ru(L)Cl(CO)(PPh3)2] (3) and [Ru(L)Cl(CO)(AsPh3)2] (4), (HL = 2,5-di(1H-pyrazol-1-yl)-1,4-benzoquinone) were synthesized, structurally characterized comprehensively by elemental analysis, IR, electronic, 1H/13C NMR spectral studies and their biological activities (DNA/protein interactions, antioxidant and cytotoxic activity studies) have been investigated and compared with that of appropriate precursor complexes [RuHCl(CO)(PPh3)3] (1), [RuHCl(CO)(AsPh3)3] (2) and the ligand LH. The crystal structures of both the new complexes 3 and 4 were determined by single crystal X-ray diffraction studies, which unveil that the central ruthenium ion adopted distorted octahedral geometry with L as a monobasic bidentate donor and the chelator was observed to undergo C–H activation at one of the ortho positions leading to the formation of a five-membered metallacycle. DNA/protein interactions of the complexes have been examined by photophysical studies, which demonstrated a non-intercalative binding mode of the complexes with DNA and the complexes strongly quench the fluorescence intensity of bovine serum albumin (BSA) through the mechanism of static quenching. The new complexes demonstrated better binding affinity with DNA/protein. The compounds were evaluated for their free radical scavenging ability involving DPPH radicals, hydroxyl radicals, nitric oxide radicals, superoxide anion radicals and hydrogen peroxide. The results showed that the new complexes 3 and 4 possess significant radical scavenging ability and outperform the standard antioxidants vitamin C and BHT. The results of in vitro cytotoxicity of the complexes tested against B16F10, HeLa, A549, MCF7 and SKOV3 cells by SRB assay were compared with the standard drug cisplatin and that the new complexes 3 and 4 showed better activity in inhibiting the growth of the cancer cells. The influence of chelation was seen in all the biological activities studied and the new ruthenium chelates 3 and 4 were found to be superior to the precursor complexes 1, 2 and the ligand LH in DNA/protein binding properties, radical scavenging ability and cytotoxicity.
 Please wait while we load your content...
Please wait while we load your content...

