
In situ generation of dihydropyridine for the enantioselective transfer hydrogenation of 1,4-benzoxazines†
Alexandre Aillerie,
Cyrille Gosset,
Clément Dumont,
Valentin Skrzypczak,
Philippe Champetter,
Sylvain Pellegrini,
Till Bousquet* and
Lydie Pélinski*
Université de Lille, CNRS, ENSCL, UMR 8181 – UCCS – Unité de Catalyse et de Chimie du Solide, F-59000 Lille, France. E-mail: till.bousquet@univ-lille1.fr; lydie.pelinski@univ-lille1.fr
First published on 27th May 2016
Abstract
A new strategy for the enantioselective transfer hydrogenation of benzoxazines involving an in situ generation of Hantzsch ester has been developed. Dihydroadducts were isolated in good yields (75–99%) and enantioselectivities (89–96% ee).
Asymmetric reduction of C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bonds is one of the most useful strategies for the preparation of optically active amines.1 In an international context oriented towards the development of eco-friendly reactions, organocatalytic transfer hydrogenation has emerged as a very powerful and promising tool to access a broad range of optically active compounds.2,3 This reaction is based on a biomimetic approach, using for instance organic hydride donors. For this purpose, Hantzsch dihydropyridine esters and its derivatives were particularly studied in combination with chiral Brønsted acids.4 In order to improve the ecological and the economical impact of this process, several solutions have been proposed. Among them, an asymmetric transfer hydrogenation of benzoxazines was recently developed in aqueous conditions5 and even more recently, an interesting bio-inspired asymmetric hydrogenation with the regeneration of a catalytic amount of Hantzsch ester was reported.6
N double bonds is one of the most useful strategies for the preparation of optically active amines.1 In an international context oriented towards the development of eco-friendly reactions, organocatalytic transfer hydrogenation has emerged as a very powerful and promising tool to access a broad range of optically active compounds.2,3 This reaction is based on a biomimetic approach, using for instance organic hydride donors. For this purpose, Hantzsch dihydropyridine esters and its derivatives were particularly studied in combination with chiral Brønsted acids.4 In order to improve the ecological and the economical impact of this process, several solutions have been proposed. Among them, an asymmetric transfer hydrogenation of benzoxazines was recently developed in aqueous conditions5 and even more recently, an interesting bio-inspired asymmetric hydrogenation with the regeneration of a catalytic amount of Hantzsch ester was reported.6
Besides this, multicomponent and cascade reactions offer a straightforward route to generate complexity and diversity in a single operation.7 These processes are particularly interesting for economical, synthetic and environmental points of view. In 2009, when describing the first use of benzothiazoline as organic hydride source for the organocatalytic transfer hydrogenation of imines, Akiyama also reported a three-component hydrogenation protocol wherein the benzothiazoline was formed in situ.2d
In this context and as part of our ongoing efforts in the development of multicomponent reactions8 and asymmetric transfer hydrogenations,9 we wish to report herein the first enantioselective transfer hydrogenation of benzoxazines involving an innovative in situ generation of Hantzsch ester (Scheme 1).
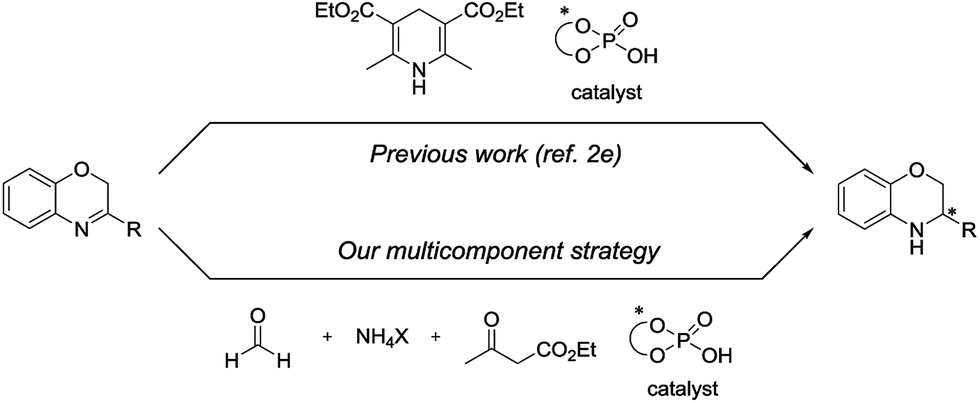 | ||
| Scheme 1 New multicomponent strategy for the transfer hydrogenation of benzoxazines. | ||
In a first set of experiments, our attention was focused on determining the appropriate source of ammonium between the acetate and the bicarbonate salts for the transfer hydrogenation of the benzoxazine 1a. For this purpose, these comparative reactions were performed in the absence of catalyst. We decided to consider ethyl acetoacetate and aqueous formaldehyde as reaction partners. Furthermore, based on Rueping's benzoxazine transfer hydrogenation, toluene was selected as solvent.2e As anticipated, when the ammonium acetate was used, the benzoxazine 1a was fully converted into the 1,2-dihydrobenzoxazine 2a while the use of ammonium bicarbonate did not allow any conversion (Table 1, entries 1 vs. 2). Indeed, if in both cases, the expected Hantzsch ester was formed in situ,10 when the acetate salt was used, the Hantzsch ester generation was accompanied by the formation of acetic acid which promoted the further transfer hydrogenation. With the aim of achieving the reaction in a catalytic and asymmetric fashion, this competitive acetic acid-promoted side reaction should be avoided. As such, ammonium bicarbonate which leads to the release of water and carbon dioxide was adopted in our further investigations.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Ammonium salt | Solvent | Time | Yieldb (%) | eec (%) |
| a Experimental conditions: benzoxazine 1a (0.24 mmol), formaldehyde (0.53 mmol, 37 wt% aq.), NH4OAc or NH4HCO3 (0.86 mmol), ethyl acetoacetate (1.22 mmol), catalyst 3 (5 mol%), solvent (2 mL), 70 °C.b NMR yield with 1,3,5-trimethoxybenzene as an internal standard.c Determined by HPLC analysis using a chiral stationary phase.d Racemic mixture of catalyst 3a.e 10 mol% catalyst loading.f Methyl tert-butyl ether.g Dimethyl carbonate.h Isolated yield.i Reaction conducted at 50 °C.j Reaction conducted at 60 °C.k Reaction conducted at 90 °C. | ||||||
| 1 | None | NH4OAc | Toluene | 24 | 95 | — |
| 2 | None | NH4HCO3 | Toluene | 24 | 0 | — |
| 3 | 3ad | NH4HCO3 | Toluene | 24 | 54 | — |
| 4 | 3ad | NH4HCO3 | Toluene | 24 | 78e | — |
| 5 | 3ad | NH4HCO3 | Toluene | 48 | 94 | — |
| 6 | 3b | NH4HCO3 | MTBEf | 48 | 76 | 72 |
| 7 | 3b | NH4HCO3 | DMCg | 48 | 78 | 70 |
| 8 | 3b | NH4HCO3 | CH3CN | 48 | 75 | 55 |
| 9 | 3b | NH4HCO3 | CH2Cl2 | 48 | 70 | 75 |
| 10 | 3b | NH4HCO3 | CHCl3 | 48 | 77 | 77 |
| 11 | 3b | NH4HCO3 | THF | 48 | 59 | 72 |
| 12 | 3b | NH4HCO3 | Toluene | 48 | 92 (87)h | 78 |
| 13i | 3b | NH4HCO3 | Toluene | 48 | 37 | 80 |
| 14j | 3b | NH4HCO3 | Toluene | 48 | 63 | 81 |
| 15k | 3b | NH4HCO3 | Toluene | 24 | 70 | 81 |
| 16 | 3c | NH4HCO3 | Toluene | 48 | 38 | 17 |
| 17 | 3d | NH4HCO3 | Toluene | 48 | 77 | 91 |
| 18 | 3e | NH4HCO3 | Toluene | 48 | 95 | 36 |
| 19 | 3f | NH4HCO3 | Toluene | 48 | 99 (93)h | 94 |
| 20 | 3f | NH4OAc | Toluene | 48 | 98 | 84 |

The first acid-promoted transfer hydrogenation was performed at 70 °C for 24 h with 5 mol% of the racemic BINOL phosphoric acid 3a. In these conditions, the 1,2-dihydrobenzoxazine was obtained in 54% yield (entry 3). While the reactivity was improved with 10 mol% catalyst loading (entry 4), the best conditions were achieved when the reaction was conducted for 48 h with 5 mol% catalyst loading, providing 2a in 94% yield (entry 5).
Subsequently, the solvent screening was conducted in the presence of a catalytic amount of the previously highlighted chiral 3,3′-phenanthryl BINOL phosphoric acid 3b.2e In this study, although most of the solvents provided the product in good conditions in respect of reactivity and selectivity (entries 6–12), the toluene appeared to be the most promising solvent with 87% isolated yield and 78% ee (entry 12).11 It is worth noticing that varying the temperature to 50, 60 or 90 °C did not significantly improve the selectivity as expected, but led to reduce the yield (entries 13–15).
Although this innovative process of in situ generation of dihydropyridine has proven his efficiency for the hydrogen transfer reduction of benzoxazines, the asymmetric induction was still disappointing under these conditions. Indeed, while 78% ee was measured on dihydrobenzoxazine 2a with our strategy, Rueping reported a 96% ee when the dihydropyridine was preformed.2e
To further increase the selectivity, we then turned our attention to diversely substituted BINOL phosphoric acids 3c–f (entries 16–19). Satisfyingly, among them, the 3,3′-bis(9-anthracenyl) derivative 3f proved to be the most effective catalyst as it increased not only the yield (93%) but also substantially the selectivity to 94% ee (entry 19).
Interestingly, the enantioselectivity of the reaction remained high when the alternative ammonium acetate was used (84% ee, entry 20). Knowing that the in situ generated acetic acid was able to promote the racemic formation of 2a in competition with the asymmetric pathway, such a high selectivity might be surprising and has to be noticed.
Although no improvement on the enantioselectivity was observed when diverse Hantzsch esters were evaluated,12 these trials revealed one practical aspect of our method. Indeed, for this study, evaluating different dihydropyridines for the transfer hydrogenation just required the addition of different acetoacetates.
The mechanism relative to the in situ Hantzsch ester generation for transfer hydrogenation of benzoxazine 1a is proposed in Scheme 2. The multicomponent reaction involving ethylacetoacetate, ammonium bicarbonate and formaldehyde leads to the formation of the dihydropyridine. As soon as it was formed, the latter reacts with the catalyst-activated benzoxazine, furnishing thus the desired 1,2-dihydrobenzoxazine. Interestingly, although it was proven that polar solvents such as acetic acid, water or alcohols were more adequate for the synthesis of Hantzsch esters,13 after choosing toluene as solvent, the NMR spectrum of the crude reaction showed the full formation of Hantzsch and its oxidized form.14 As an explanation, it might be assumed that in addition to beneficial presence of the Brønsted acid catalyst, the generation of the dihydropyridine was favoured since it was consumed into its pyridine analog as it was formed.
 | ||
| Scheme 2 Reaction mechanism. | ||
With the optimized conditions in hand, the scope of the multicomponent transfer hydrogenation was extended to the formation of diversely substituted dihydrobenzoxazines (Table 2). For this purpose, several benzoxazines were synthesized by condensation of bromoacetophenones with 2-aminophenol derivatives and engaged in the reaction at 70 °C, in toluene and in the presence of NH4HCO3, formaldehyde and ethyl acetoacetate and 5 mol% of phosphoric acid 3c.15
| Entry | Reagent | Product | Yieldb (%) | eec (%) | |
|---|---|---|---|---|---|
| a Experimental conditions: benzoxazine 1 (0.24 mmol), 37% aq. formaldehyde (0.53 mmol), NH4HCO3 (0.86 mmol), ethyl acetoacetate (1.22 mmol), 3f (0.012 mmol), toluene (2 mL), 70 °C, 48 h.b Isolated yields.c Determined by HPLC analysis using a chiral stationary phase. | |||||
| 1 | 1a |  |
2a | 93 | 94 |
| 2 | 1b | 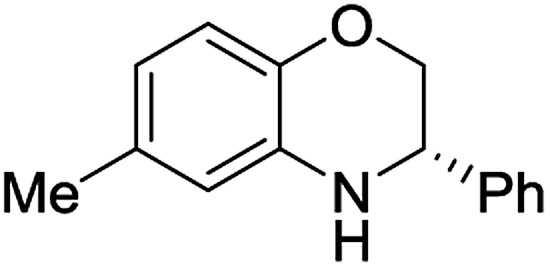 |
2b | 99 | 89 |
| 3 | 1c |  |
2c | 89 | 91 |
| 4 | 1d |  |
2d | 75 | 93 |
| 5 | 1e |  |
2e | 99 | 89 |
| 6 | 1f |  |
2f | 98 | 96 |
| 7 | 1g |  |
2g | 91 | 95 |
The dihydrobenzoxazines were isolated in a good 75–99% yield range and enantioselectivities values from 89 to 96% (entries 1–7). The best selectivities of 96 and 95% were measured for the products bearing a bulky bromo or phenyl substituent in the position 4 of the phenyl group (entries 6 and 7).
Conclusions
In summary, we have developed a transfer hydrogenation of benzoxazines where the reducing agent, the Hanztsch ester, was generated in situ in the presence of a catalytic amount of BINOL phosphoric acid 3f. The reaction allowed the formation of diverse enantioenriched dihydrobenzoxazines in good yields and enantioselectivities.Acknowledgements
Chevreul institute (FR 2638), Ministère de l’Enseignement Supérieur et de la Recherche, Région Nord – Pas de Calais and FEDER are acknowledged for supporting and funding this work. This research was supported by the “Conseil Régional Nord-Pas de Calais” (program PRIM, grant for AA).Notes and references
- T. C. Nugent, Chiral Amines Synthesis: Methods, Developments and Applications, Wiley-VCH, Germany, 2010 Search PubMed.
- (a) M. Rueping, E. Sugiono, C. Azap, T. Theissmann and M. Bolte, Org. Lett., 2005, 7, 3781 CrossRef CAS PubMed; (b) S. Hoffmann, A. M. Seayad and B. List, Angew. Chem., Int. Ed., 2005, 44, 7424 CrossRef CAS PubMed; (c) R. I. Storner, D. E. Carrera, Y. Ni and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 84 CrossRef PubMed; (d) C. Zhu and T. Akiyama, Org. Lett., 2009, 11, 4180 CrossRef CAS PubMed; (e) M. Rueping, A. P. Antonchick and T. Thiessmann, Angew. Chem., Int. Ed., 2006, 45, 6751 CrossRef CAS PubMed; (f) M. Rueping, A. P. Antonchick and T. Thiessmann, Angew. Chem., Int. Ed., 2006, 45, 3683 CrossRef CAS PubMed; (g) Z. Zhang and H. Du, Org. Lett., 2015, 17, 6266 CrossRef CAS PubMed; (h) G. Li, H. Liu, G. Lv, Y. Wang, Q. Fu and Z. Tang, Org. Lett., 2015, 17, 4125 CrossRef CAS PubMed; (i) C. Bleschke, J. Schmidt, D. S. Kundu, S. Blechert and A. Thomas, Adv. Synth. Catal., 2011, 353, 3101 CrossRef CAS; (j) D. S. Kundu, J. Schmidt, C. Bleschke, A. Thomas and S. Blechert, Angew. Chem., Int. Ed., 2012, 51, 5456 CrossRef CAS PubMed; (k) Z. Zhang, Y. R. Ji, L. Wojtas, W.-Y. Gao, S. Ma, M. J. Zaworotko and J. C. Antilla, Chem. Commun., 2013, 49, 7693 RSC; (l) Z.-P. Chen, M.-W. Chen, R.-N. Guo and Y.-G. Zhou, Org. Lett., 2014, 16, 1406 CrossRef CAS PubMed; (m) Y. Zhang, R. Zhao, R. L.-Y. Bao and L. Shi, Eur. J. Org. Chem., 2015, 3344 CrossRef CAS.
- For reviews see: (a) M. Rueping, J. Dufour and F. R. Schoepke, Green Chem., 2011, 13, 1084 RSC; (b) D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621 CrossRef CAS PubMed; (c) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef CAS PubMed.
- C. Zheng and S.-L. You, Chem. Soc. Rev., 2012, 41, 2498 RSC.
- M. Rueping and T. Theissmann, Chem. Sci., 2010, 1, 473 RSC.
- Q.-A. Chen, M.-W. Chen, C.-B. Yu, L. Shi, D.-S. Wang, Y. Yang and Y.-G. Zhou, J. Am. Chem. Soc., 2011, 133, 16432 CrossRef CAS PubMed.
- (a) J. Zhu, Q. Wang and M. Wang, Multicomponent Reactions in Organic Synthesis, Wiley-VCH, Germany, 2015 Search PubMed; (b) L. F. Tietze, H. P. Bell and G. Brasche, Domino Reactions in Organic Synthesis, Wiley-VCH, Germany, 2006 Search PubMed; (c) H. Pellissier, Asymmetric domino reactions, RSC. Catalysis Series, Cambridge, 2013 Search PubMed.
- S. Pellegrini, J.-N. Grad, T. Bousquet and L. Pélinski, Tetrahedron Lett., 2011, 52, 1742 CrossRef CAS.
- A. Aillerie, V. L. de Talancé, A. Moncomble, T. Bousquet and L. Pélinski, Org. Lett., 2014, 16, 2982 CrossRef CAS PubMed.
- As observed by 1H-NMR of the crude reactions.
- Absolute configuration was determined by comparison with the data in ref. 2e.
- Results not reported here. For this evaluation, the dihydropyridines were generated in situ from the benzyl, isopropyl or allyl acetoacetates.
- (a) A. Hantzsch, Justus Liebigs Ann. Chem., 1882, 215, 1 CrossRef; (b) F. Tamaddon, Z. Razmi and A. A. Jafari, Tetrahedron Lett., 2010, 51, 1187 CrossRef CAS; (c) J. Xia and G. Wang, Synthesis, 2005, 2379 CAS; (d) D. M. Stout and A. I. Meyers, Chem. Rev., 1982, 82, 223 CrossRef CAS.
- NMR estimation with 1,3,5-trimethoxybenzene as internal standard shows signals relative to Hantzsch ester and its oxidized analog.
- J. L. Núñez-Rico and A. Anton Vidal-Ferran, Org. Lett., 2013, 15, 2066 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra04930c |
| This journal is © The Royal Society of Chemistry 2016 |