
RuO2@Co3O4 heterogeneous nanofibers: a high-performance electrode material for supercapacitors†
Abstract
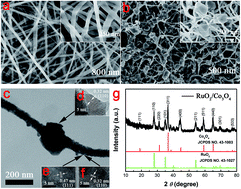
Novel RuO2@Co3O4 heterogeneous nanofibers (HNFs) were synthesized by a simple electrospinning method, followed by calcination. The possible mechanism of formation of RuO2@Co3O4 HNFs is presented. The synergy between RuO2 and Co3O4 and the unique feature of heterostructure allow the composite to exhibit a high reversible capacity of 1103.6 F g−1 at a current density of 10 A g−1 and a high-rate capability of 500.0 F g−1 at a current density of 100 A g−1. Moreover, excellent cycling performance was observed, where the capacities could be maintained at 93.0%, 91.8%, and 88.0% after 1000, 2000 and 5000 cycles, respectively, at a current density of 10 A g−1. This study therefore confirms that the as-prepared RuO2@Co3O4 HNFs can serve as advanced supercapacitor (SCs) materials. It is highly anticipated that this simple method of electrospinning can be extended to preparing other new types of heterostructural materials that could be used in the field of electrochemical energy storage.
 Please wait while we load your content...
Please wait while we load your content...

