
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microwave-assisted hydrothermal synthesis and spectroscopic characteristics of a Lu4Hf3O12:Pr scintillator†
J.
Trojan-Piegza
* and
E.
Zych
Faculty of Chemistry, University of Wroclaw, 14 F. Joliot-Curie Street, 50-383 Wroclaw, Poland. E-mail: joanna.trojan-piegza@chem.uni.wroc.pl
First published on 1st June 2016
Abstract
Exclusively high density of Lu4Hf3O12 makes it an attractive host for scintillator materials. This paper presents the versatility of microwave-assisted hydrothermal technology (MAH) to fabricate nanocrystalline Lu4Hf3O12:Pr luminescent powder. It is shown that by varying the parameters of the fabrication procedure, changing the composition of the liquid medium and/or using polyethylene glycol, PEG 2000, as a surfactant, the morphology of the powders could be modified. In the presence of PEG, obtaining non-agglomerated particles ∼5 nm in diameter was possible. Sintered ceramics at 1700 °C were fabricated using various powders. Photoluminescence and X-ray excited emission spectra showed the Pr3+ emission resulting from 4f → 4f transitions. At room temperature the 3P0 and 1D2 levels contributed roughly equally to the total emission in both the powders and sintered ceramics. The former produced luminescence with a decay time of ∼15 μs while the latter was an order of magnitude slower. At 10 K the emission resulted almost exclusively from the 3P0 level. In addition, a broad-band blue luminescence was observed at low temperatures. Room temperature radioluminescence (RL) was found to be rather inefficient in both the powders and ceramics.
1. Introduction
Lutetium hafnate, Lu4Hf3O12, with a high density, 9.9 g cm−3, and the presence of heavy atoms Hf and Lu, provides high stopping power for X- and γ-rays and ensures high photofraction – the ability to stop/absorb a high-energy particle in one position due to photoelectric absorption rather than in a few spatially separated locations by means of Compton scattering.1,2 Consequently, Lu4Hf3O12 is potentially a good matrix for scintillation detectors. In the past Lu4Hf3O12 doped with 0.5% of Eu3+, Tb3+, Pr3+ or Bi3+ was prepared by solid-state technology and some scant photoluminescence and radioluminescence data were reported.3 More details on the spectroscopic properties of Eu3+- and Tb3+-activated Lu4Hf3O12 prepared by a combustion method were published by Mihokova et al.4 and Lauria et al.5It was shown that controlling the material’s morphology allows for the management of their ferroelectric/dielectric,6,7 electrochemical8 or photoluminescence (PL)9 properties. Hydrothermal technology presents many benefits for nanomaterial preparation. Chemical homogeneity of the products, moderate temperatures and simplicity are among them. Combining microwaves with the hydrothermal technique offers more advantages including much shorter preparation time and prompt, effective crystallization.10,11 Consequently, the process is also energy- and time-efficient.
Good control of grain shape and agglomeration as well as a narrow crystallite size distribution were reported for microwave-assisted processes.12 It was also shown that by varying the temperature, pressure and duration of the MAH process as well as the pH of the initial solution, one can significantly influence the product morphology and physicochemical properties.13–15
The aim of the present study is to evaluate microwave-assisted hydrothermal synthesis (MAH) as a preparation technique of Lu4Hf3O12:Pr powders and to characterize their luminescence properties. Thus, the influence of the process parameters, pH of the solution and composition of the solvent on the morphology of the Lu4Hf3O12:Pr powders as well as their optical characteristics is discussed in detail. The Pr activator was chosen to determine whether it can produce fast 3P0 luminescence, which is useful in medical imaging, mostly in computed tomography (CT). Such an emission from the very dense host of Lu4Hf3O12 could be of special interest as it is a competitor for Gd2O2S:Pr used in CT machines nowadays. Therefore, sintered ceramics of Lu4Hf3O12:Pr were also fabricated and their luminescence properties were examined among others under X-ray and VUV-UV synchrotron radiation. Among others, time-resolved spectroscopy at room and helium temperatures was employed.
2. Experimental details
Lu2O3, Pr6O11, and HfCl4 reagents (Stanford Materials 99.999%) were used as starting chemicals. Their stoichiometric mixtures were dissolved in nitric acid, evaporated and 1 M KOH water solution was added dropwise until pH = 9 was achieved. The raw precipitate was filtered, washed with water and placed in a PTFE reaction vessel of a microwave reactor (ERTEC, Poland) that works at 2.45 GHz with a maximum power of 600 W and temperature up to 300 °C. The process was controlled by programming the power and/or temperature and time. The precipitate was treated in the microwave reactor in three different reaction media: water, KOH solution of pH = 9 or ethylene glycol (EG). A series of experiments was performed with the additional use of polyethylene glycol, PEG 2000 (polyethylene glycol of molecular weight 1900–2200), as a surfactant. The PEG amount was 1%, 5% or 50% of the mass of the precipitate placed in the vessel. The microwave reactions were carried out for different periods of time (20, 40, 80, and 160 minutes), at different temperatures (140, 160, 200, and 240 °C) and with various fluid (suspension) volumes (30, 50, and 75 cm3). The parameter profiles of a typical microwave-assisted hydrothermal synthesis of the Lu4Hf3O12:Pr nanopowders are presented in Fig. 1. After microwave treatment the products were filtered, washed several times and dried at 80 °C. The powders were also heat-treated at 700 °C or 1000 °C. For spectroscopic measurements a nominal Pr content of 0.01, 0.05, 0.25, 0.75, 1.25, 2.5 and 5 mol% with respect to Lu was used. Portions of the powders, after cold-pressing, were sintered at 1700 °C for 5 h under vacuum. Despite the quite different morphologies of the raw powders, the sintering proceeded quite similarly.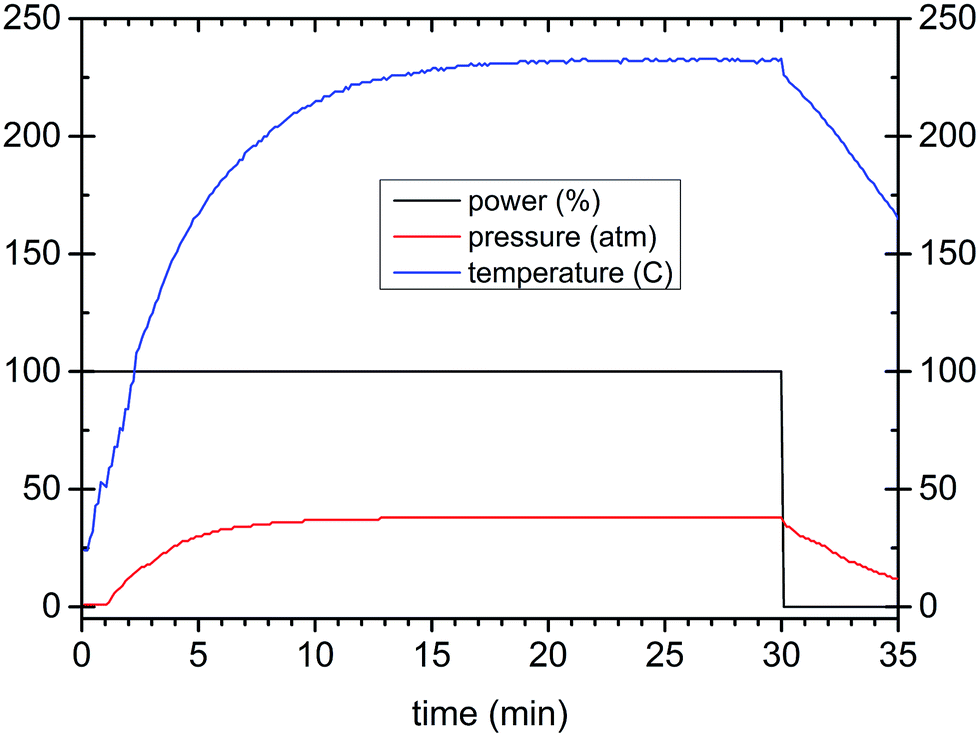 | ||
| Fig. 1 A parameter profile demonstration of a typical MAH synthesis of Lu4Hf3O12. | ||
The crystallinity and crystallographic purity of the materials were analyzed with a Bruker D8 Advance diffractometer using Ni-filtered Cu Kα1 (1.54056 Å) radiation. The morphology of the specimens was examined with an FEI Tecnai G2 20 X-TWIN transmission electron microscope (TEM) and a Hitachi S-3400N scanning electron microscope (SEM) equipped with an energy dispersive X-ray spectroscopy (EDX) analyzer. Photoluminescence measurements were performed under synchrotron radiation at the Superlumi station at DESY HasyLab in Hamburg. Lifetime measurements upon 280 nm excitation were performed with an FLS 980 Spectrofluorimeter from Edinburgh Instruments equipped with a 60 W pulse Xe lamp and an S900 photomultiplier detector operating within 180–870 nm. Radioluminescence spectra were recorded with a setup consisting of an HR2000 CG spectrometer with a CCD detector coupled to a QP-2-600-2-SR-BX optical fiber equipped with a 74 UV collimating lens. The resolution of the system was about 1.2 nm. Unfiltered white X-rays from a Cu X-ray tube operating at 40 kV were used to excite the specimens.
3. Results and discussion
3.1 X-ray diffraction
The lutetia–hafnia system investigated in this paper crystallizes as a Lu4Hf3O12 phase in a rhombohedral structure with an R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) h space group. This is derived from a fluorite-type structure with a 3a metal site occupied by Hf in octahedral symmetry and 18f metal position shared by hafnium and lutetium in a ratio of (Hf + 2Lu)/3.16 Thus lutetium hafnate crystallizes in the not fully ordered phase which should impact its PL spectra, broadening the spectral features.
h space group. This is derived from a fluorite-type structure with a 3a metal site occupied by Hf in octahedral symmetry and 18f metal position shared by hafnium and lutetium in a ratio of (Hf + 2Lu)/3.16 Thus lutetium hafnate crystallizes in the not fully ordered phase which should impact its PL spectra, broadening the spectral features.
X-ray diffraction studies presented in Fig. 2 reveal that the pure Lu4Hf3O12 phase is formed already after the MAH reaction was carried out at 160 °C for 20 minutes. Extension of the MAH processing time up to 200 minutes causes only a slight increase of the intensities of the diffraction lines (not shown). As Fig. 2 shows, the diffraction lines are very broad, but well-defined and distinct, indicating that the powder is well crystallized though presumably it consists of very small crystallites. After the heat-treatment at 700 °C of the as-made precipitate, the XRD patterns are hardly changed. Further heating at 1000 °C and 1700 °C leads to a drastic narrowing of the diffraction lines indicating a significant enlargement of the crystallites and consequently a substantial mass transfer between the grains. Also after various heat-treatments only the pure Lu4Hf3O12 phase is identified as the product.
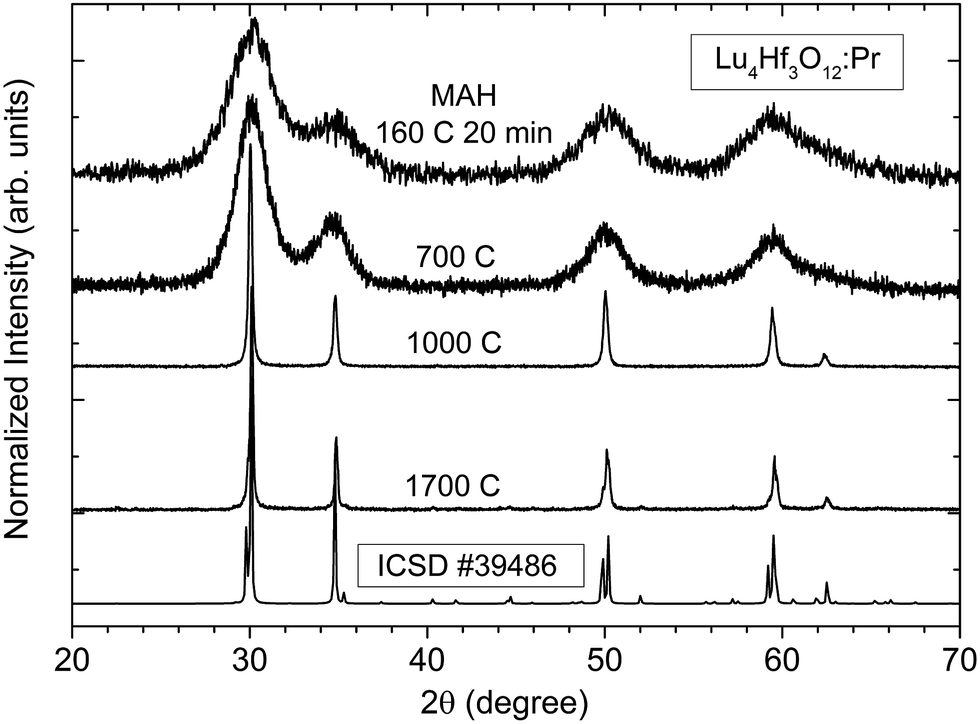 | ||
| Fig. 2 XRD patterns of the Lu4Hf3O12 as-made, post-treated powders and ceramics. | ||
3.2 Morphology
TEM examination, see Fig. 3a, reveals that the raw powders synthesized in water consist of spherical crystallites a few nanometers in diameter and are very uniform both in size and shape. Heat-treatment at 700 °C (Fig. 3b) only slightly enlarges the size of the crystallites, retaining their shapes. Heating at 1000 °C (Fig. 3c) causes a noticeable growth of the crystallites to about a few tens of nm and causes their partial sintering and aggregation. Fig. 3d presents typical SEM images of the ceramics sintered at 1700 °C. Quite uniform distribution of the grain sizes is revealed, and most of the grains are about 2–3 μm large. No voids between or within the grains and triple points are seen. Thus, the nanoparticles of Lu4Hf3O12 prepared by means of microwave-assisted hydrothermal synthesis are sintered efficiently. On the other hand, the pellets do not show any noticeable transparency, which indicates that preparing high-quality sintered pellets of Lu4Hf3O12:Pr, if needed, would require a significant number of experiments to optimize the procedure. | ||
| Fig. 3 TEM images of Lu4Hf3O12 synthesized by the MAH method in water solution: (a) raw powder, (b) heat treated at 700 °C, (c) 1000 °C and (d) typical SEM image of all ceramics fabricated at 1700 °C. The insets show the SAED images. | ||
 | ||
| Fig. 4 TEM images of the ethylene glycol-medium MAH synthesized Lu4Hf3O12 powder. Right and left images present the same powder at different magnifications. | ||
 | ||
| Fig. 5 TEM images of the alkaline-medium (pH ∼ 9) MAH synthesized Lu4Hf3O12 powder. Right and left images present the same powder at different magnifications. | ||
The mechanism of the observed changes of the morphology was not investigated in detail. However, there are numerous papers which raise the problem in oxide nanopowders. A great example of how the concentration and chemical composition of the water solution/suspension may influence the final product morphology is given by Wang et al.17 By varying the processing parameters the authors were able to obtain Lu2O3:Eu nanocrystalline powders in the form of nanoflakes, thin and long nanorods, and nanoquadrels. Significant variations in morphology were also achieved for Gd2O3 – hollow or spherical particles were obtained, with sizes varying from hundreds of nanometers to microns.18 Fan et al.19 showed, for LaVO4, the influence of the pH of suspension and time of synthesis on the final product morphology. Thus, it is not a surprise that from the solution containing KOH the powder of Lu4Hf3O12 is obtained with different particle sizes and shapes. The EDX measurements prove that the powders synthesized at pH = 9 contain quite a significant amount of K+ ions presumably adsorbed on the surface of the grains/crystallites (see Fig. SI-1†). Apparently, OH− ions also adsorb on the particles. All this may affect the crystallization process, altering the thermodynamic and/or kinetic parameters of the Lu4Hf3O12 crystallization.
 | ||
| Fig. 6 TEM images of the MAH synthesized Lu4Hf3O12 powder with the addition of PEG for hydroxide precipitation in indicated amounts: 1% (a), 5% (b), 50% (c). Right and left images present the same powder with different magnifications. | ||
3.3 Photoluminescence
All of the as-made powders hardly show any luminescence, which is quite typical for materials made in water, as they contain either water molecules or OH groups which effectively quench luminescence by a multiphonon nonradiative relaxation. After heat-treatment at 700 °C the luminescence can be easily recorded independent of the preparation procedure. Applying higher temperatures further improves the PL intensity and this is observed for all of the powders. The ceramics sintered using different powders show a very similar morphology (see Fig. 3d) and perform comparably in terms of PL and RL. Their spectra are practically indistinguishable. This may be taken as proof that the high-temperature treatment ends up with spectroscopically undifferentiated materials with very similar crystallinity, microstructure, and dopant dispersion within the host as well as defect types and concentrations.For all heat-treated powders as well as sintered ceramics only the emission features related to the Pr3+ ions are observed and luminescence from both the 3P0 and 1D2 levels is seen.
Fig. 7 shows RT, time-resolved emission and (averaged) excitation spectra of the Lu4Hf3O12:Pr ceramics with two different concentrations of dopant: 0.01% and 0.75%. Both specimens show intense PL emissions. The excitation spectra at 611 nm luminescence consist of a broad band peaking at 280 nm and originating presumably from the intraconfigurational 4f → 5d transition of Pr3+. PL emissions upon excitation into the 280 nm band result exclusively from the intraconfigurational 4f → 4f transitions of the Pr3+ ions. The time resolved luminescence measurements allow us to identify with confidence the features resulting from the relaxation of the 3P0 and 1D2 levels. The former decays much faster as the 3P0 → 3H4 transition is spin allowed, while the latter is forbidden. It is thus clear that around 600–650 nm emissions from both levels are generated and they strongly overlap. In the blue-green part of the spectrum only the emission from 3P0 contributes to the luminescence and these features practically disappear about 170 μs after the excitation pulse. It is striking that the evolution of the emission in time is very similar for both Pr concentrations (0.01% and 0.75%). The slow decaying luminescence from the 1D2 level contributes about 30% to the total PL intensity. Thus it can be concluded that at RT the interaction of the activator ions at the higher Pr content does not facilitate the 3P0 → 1D2 nonradiative relaxation and especially the cross-relaxation does not participate in the depopulation of the 3P0 level. Consequently, it is rather a 3P0 → 1D2 multiphonon nonradiative relaxation which feeds the 1D2 level. If that conclusion is true, lowering the temperature, and thus reducing the effectiveness of the host lattice vibrations in bridging the 3P0 and 1D2 levels should enhance the relative intensity of the luminescence from the 3P0 level. The lack of a d–f emission of the Pr3+ ions (also at low temperatures as will be presented later) suggests the immersion of the lowest 5d level within the host lattice conduction band.20 It is noteworthy that the f–f emission features are relatively broad. This effect reflects the disordered structure of the Lu4Hf3O12 host, as already anticipated in Section 3.1.
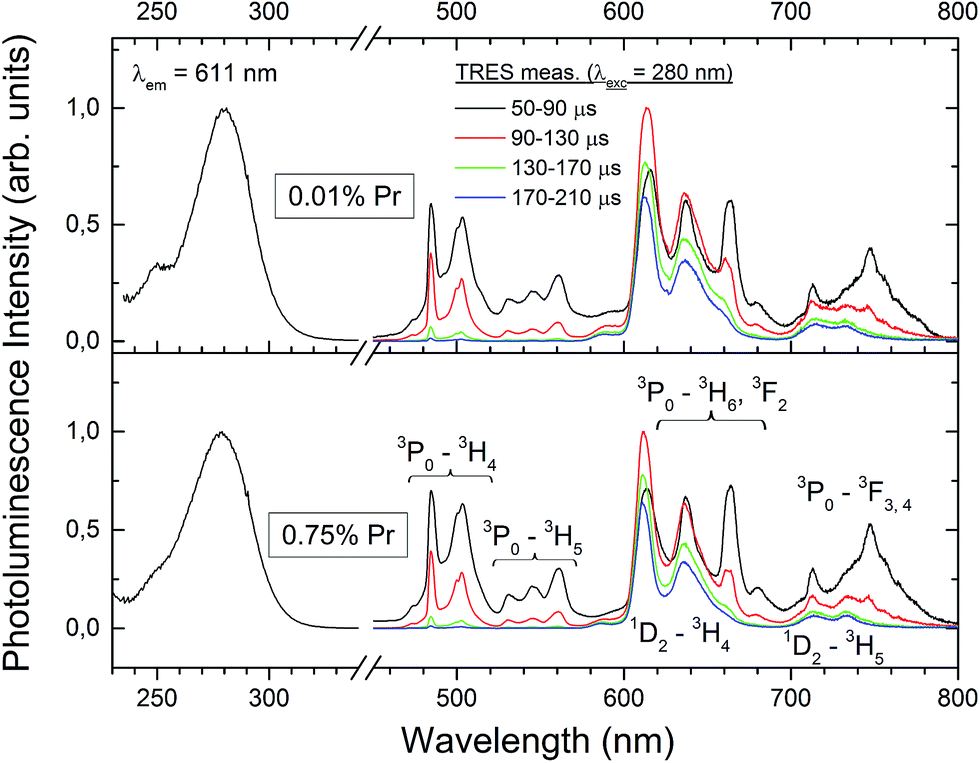 | ||
| Fig. 7 Room temperature PL excitation and time-resolved emission spectra of the Lu4Hf3O12:Pr sintered ceramics. | ||
Fig. 8a presents the time resolved luminescence spectra of the 0.75% ceramic specimen (compare to RT spectra in Fig. 7) recorded at 20 K upon excitation at 280 nm and normalized at 611 nm. The normalization is done where the contribution from the slow 1D2 emission is dominant compared to the 3P0 luminescence. Some narrowing of the emission features is observed compared to the RT data, although the characteristics for the 4f-element line resolution do not appear even at 20 K. This agrees with the structural characteristics of the host lattice in which Lu shares the same crystallographic position with Hf. Apparently, this affects the crystal field symmetry and strength around the Pr3+ ions. Consequently, even for very low concentrations, when the Pr ↔ Pr interactions are negligible, the activator ions feel slightly different crystal fields, which are single-ion-specific rather than site-specific. The time resolved low-temperature emission spectra confirm the conclusions drawn from the RT luminescence (Fig. 7), that the luminescence from the 1D2 state appears in the range of ∼600–650 nm. At low temperature its contribution to the total emission is even lower than at RT and may be estimated as <10%. As was already mentioned, such a behaviour is expected when the 3P0 → 1D2 nonradiative relaxation occurs mostly due to multiphonon relaxation, which becomes continuously less effective as the temperature decreases. Let one note that at helium temperature the d–f luminescence is also not present which proves that the lowest 5d level is located within the host conduction band, as was stated before.
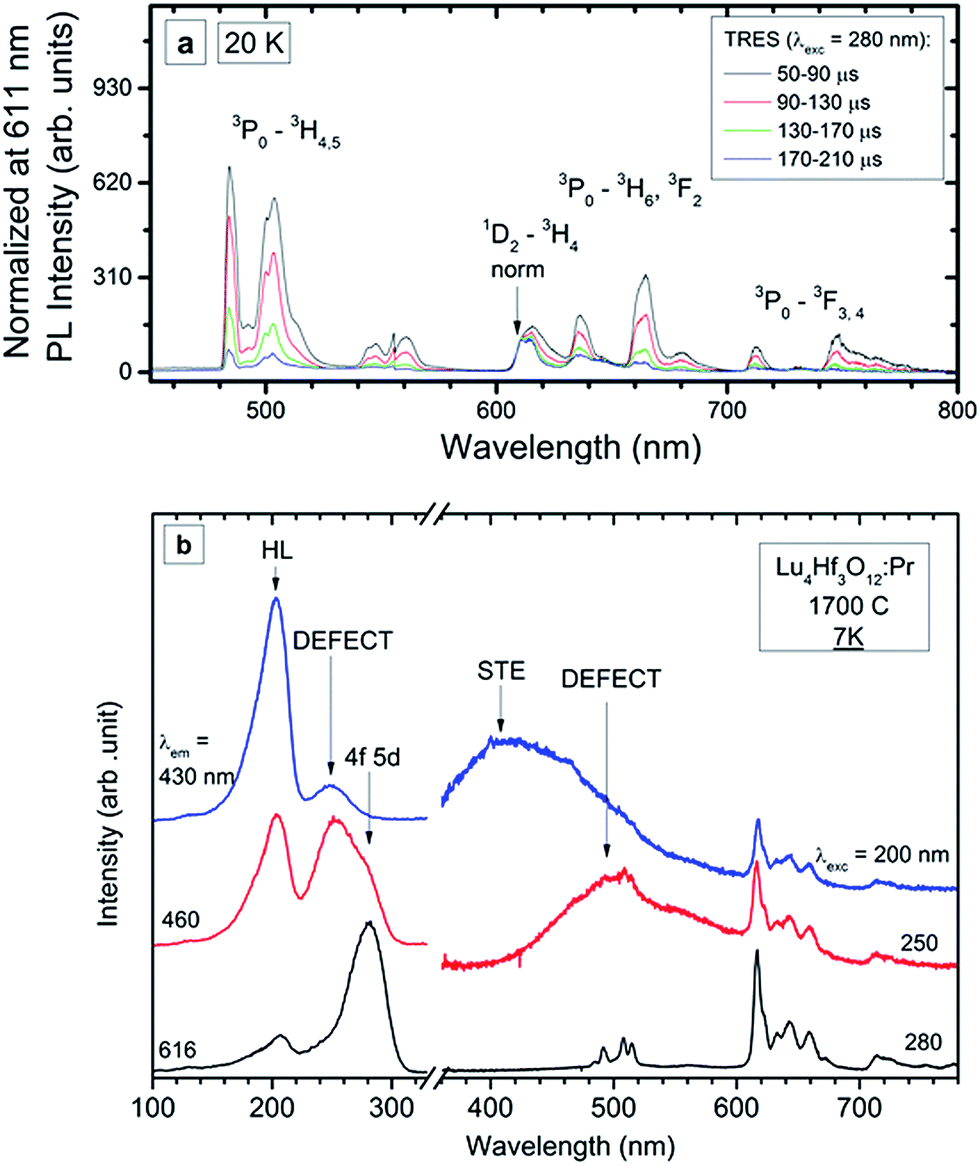 | ||
| Fig. 8 PL time-resolved emission spectra (TRES) recorded at 20 K (a) and PL emission and excitation spectra recorded at 7 K (b) of the Lu4Hf3O12:Pr 0.75% sintered ceramics. | ||
Low temperature measurements were also performed for the Lu4Hf3O12:Pr 0.75% ceramics using high-energy synchrotron excitation – in the short UV and VUV range of the spectrum. The experimental data are presented in Fig. 8b. The spectra reveal at least two new luminescence features – broad band emissions in the violet and blue-green parts of the spectrum. The f → f luminescence of Pr3+ is present independent of the excitation energy. When changing the excitation wavelength from 280 nm to 250 nm, the Pr3+ emission is partially overlaid with a broad-band luminescence peaking around 500 nm. A comparison of the excitation spectra at 616 nm (Pr3+) and the broad band luminescence at 460 nm proves that the latter has its own excitation band peaking around 250 nm. Since this excitation feature is located below the band gap energy (∼200 nm), the band peaking at about 500 nm can be assigned to a defect state, presumably an F+ centre whose existence was reported for undoped Lu4Hf3O12.21 Generation of such defects, basically connected with oxygen vacancies, is rational due to the high-temperature fabrication of the ceramics.22
A common feature of the excitation spectra is the band peaking around 202 nm. Excitation into this component gives rise to another broad-band emission which peaks around 420–430 nm. The excitation spectrum of this luminescence recorded at 7 K is dominated by an intense band around 200 nm. In addition, a low intensity feature peaking around 250 nm is present. This seems to reflect the fact that at 430 nm defect luminescence of lower energy is detected also (see red line in Fig. 8b). Thus, the excitation of the 430 nm luminescence is composed of just the strong band around 200 nm coming from the fundamental absorption of the host lattice. Since the 430 nm luminescence can be excited only by the elevation of electrons from the valence to the conduction band and the Stokes’ shift reaches a great value of ∼27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1, it is tentatively assigned to a self-trapped exciton (STE), as this would justify such a significant relaxation after excitation and before emission. Part of the energy after excitation over the band-gap is transferred to the Pr3+ ions as their luminescence is also present then. Even under the high-flux excitation with synchrotron radiation no d → f luminescence of Pr3+ is seen.
000 cm−1, it is tentatively assigned to a self-trapped exciton (STE), as this would justify such a significant relaxation after excitation and before emission. Part of the energy after excitation over the band-gap is transferred to the Pr3+ ions as their luminescence is also present then. Even under the high-flux excitation with synchrotron radiation no d → f luminescence of Pr3+ is seen.
Fig. 9 presents the RT decay profiles of the 3P0 → 3H4 luminescence at 502 nm (Fig. 9a) and the mixed luminescence from both the 3P0 and 1D2 → 3H4 levels at 611 nm (Fig. 9b). Reasonable fits can be obtained using two components and the average time constants of both emissions (3P0 and 1D2) are also given in the graphs. The 3P0 emission decays with a radiative lifetime of 13–14 μs and does not differ for the two Pr concentrations. This agrees with the results presented in Fig. 7 which showed that the ratio of the emission from the 3P0 and 1D2 levels does not change with Pr content. Also the decay of the 1D2 luminescence does not show any concentration quenching, as the decay traces for both concentrations perfectly overlap (Fig. 9b). The first part of the decay of the 611 nm emission is fast and the fit gives a time constant of ∼15 μs in this region, which indicates that this component comes from the overlapping luminescence of the 3P0 level. The overlap is also seen in the time-resolved spectra presented in Fig. 7 and 8. Thus, the kinetic measurements perfectly support the data and conclusions presented above.
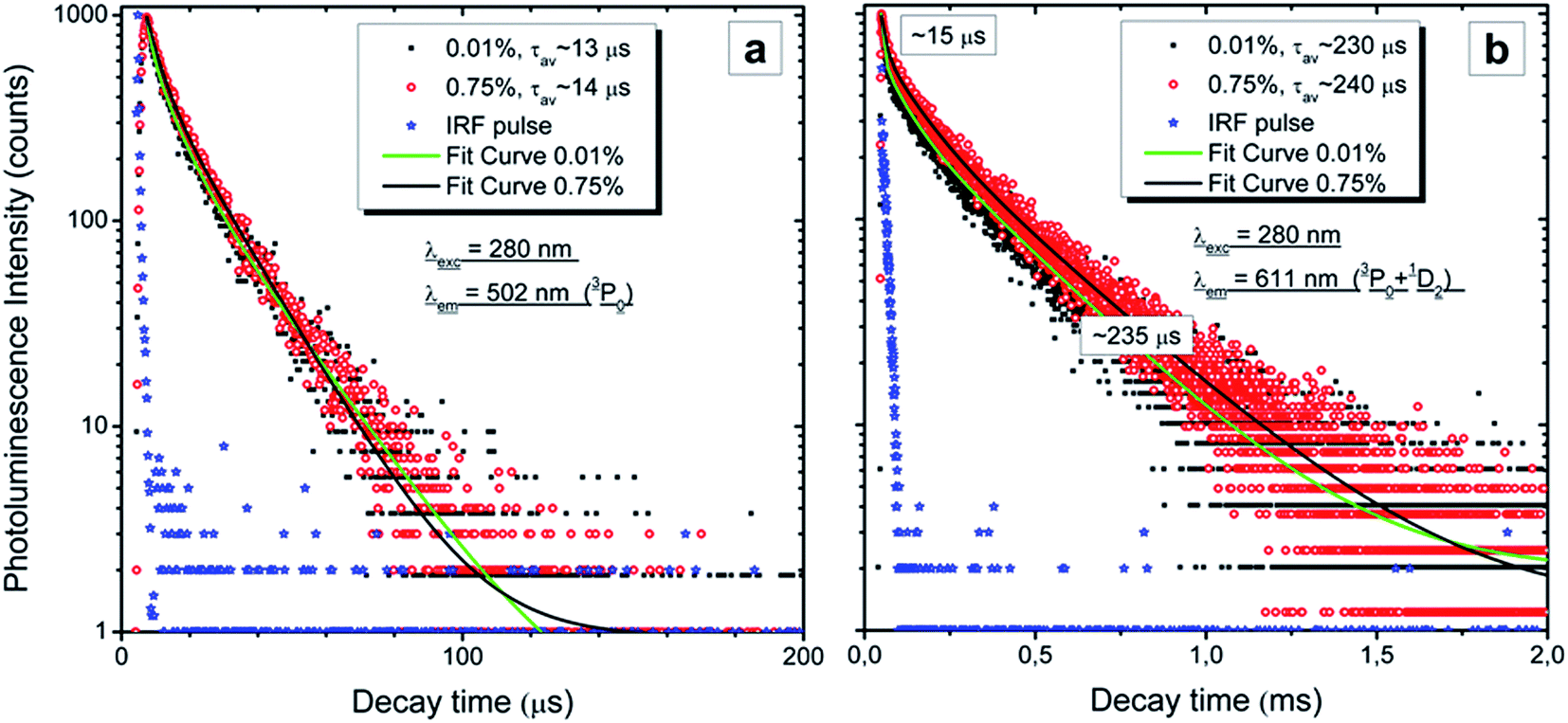 | ||
| Fig. 9 Room temperature decay curves with fits of chosen emission of the Lu4Hf3O12:Pr sintered ceramics containing a different amount of dopant. In the legends the values of the average lifetime are given. | ||
3.4 Radioluminescence
Lutetium hafnate with a high density of 9.9 g cm−3 appears to be an obvious host lattice for scintillators and X-ray phosphors as its stopping power is impressive. X-ray excited spectra were recorded for all of the Lu4Hf3O12:Pr sintered ceramics and a representative spectrum is presented in Fig. 10. Similar to the PL emission the RL originates from both the 3P0 and 1D2 levels. Unfortunately, it is disappointing that the overall RL efficiency does not exceed 10–15% of the light yield of the commercial GOS X-ray phosphor. Thus, the Pr3+ radioluminescence in this host is perceptible but faint.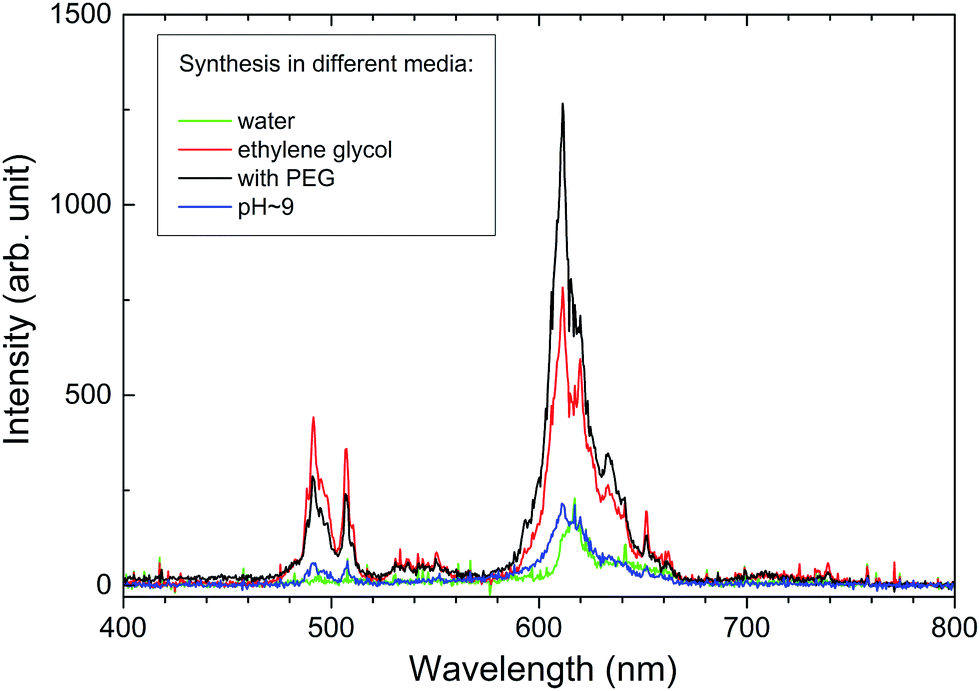 | ||
| Fig. 10 Radioluminescence spectra of the Lu4Hf3O12:0.75% Pr ceramics sintered at 1700 °C from powder MAH synthesized in different solutions: water (green), ethylene glycol (red), KOH of pH = 9 (blue), with PEG addition (black). | ||
The overall RL/scintillation process occurs in three steps.1,2 First, absorption of the high energy ionizing radiation and conversion of the incoming particle energy into a number of electron–hole pairs occurs. At this stage the host lattice gets excited. Then the energy of the free carriers is transferred to the luminescent center, Pr3+ in this case. Thus, at this stage the excited carriers diffuse toward the luminescent ions and excite them, getting sequentially trapped: Pr3+ + e + h → Pr4+ +e → (Pr3+)*. Finally, the excited dopant radiatively dissipates its energy ((Pr3+)* → Pr3+ + hν) closing the cycle. Since the last step is found to be efficient in photoluminescence experiments and the first step produces an ample number of electron–hole pairs,1,2 the intermediate step of the scintillation has to be the most deleterious for the final RL efficacy in the Lu4Hf3O12:Pr ceramics. Thus, it appears that the reason for the low RL efficiency in Pr doped Lu4Hf3O12 is presumably an inefficient energy transfer from the excited host to the emitting center.
4. Conclusions
Nanosized Pr-doped lanthanum hafnate powders, Lu4Hf3O12:Pr, with different morphologies were obtained by a microwave-assisted hydrothermal method. Reaction control allowed us to demonstrate the influence of the synthesis medium as well as the presence of surfactant on the product morphology. Independent of the synthesis route, the pure phase of rhombohedral Lu4Hf3O12 was revealed by X-ray diffraction measurements. Both the heat-treated powders of all morphologies as well as the sintered ceramics produced photo- and radioluminescence. The spectra consisted of f–f transitions of Pr3+ ions. In the sintered ceramics only, low temperature photoluminescence under synchrotron radiation revealed a broad-band emission composed of two components, which were identified as resulting from a self-trapped exciton and from a defect, presumably an F+ center. The RL efficiency appeared to be much weaker than that of a commercial GOS, apparently due to an inefficient host-to-Pr energy transfer.Acknowledgements
The financial support of the Minister of Science and Higher Education under grant #N N205 015 934 and the EU through DESY grant #II-2009-0289 EC is gratefully acknowledged.References
- A. Lempicki, A. J. Wojtowicz and C. Brecher, in Wide-Gap Luminescent Materials: Theory and Applications, ed. S. R. Rotman, Kluwer Academic Publishers, Norwell, USA, 1997, Inorganic Scintillators, pp. 235–301 Search PubMed.
- E. Zych, Inorganic Display Materials, in Handbook of Luminescence, Display Materials and Devices, ed. H. S. Nalwa and L. S. Rohwer, American Scientific Publishers, California, USA, 2003, vol. 2, pp. 251–300 Search PubMed.
- L. Havlak, P. Bohacek, M. Nikl, R. Kucerkowa and V. Studnicka, Opt. Mater., 2010, 32, 1372 CrossRef CAS.
- E. Mihokova, M. Fasoli, A. Lauria, F. Moretti, M. Nikl, V. Jary, R. Kucerkova and A. Vedda, Opt. Mater., 2011, 34, 228 CrossRef CAS.
- A. Lauria, N. Chiodini, E. Fasoli, E. Mihóková, F. Moretti, A. Nale, M. Nikl and A. Vedda, J. Mater. Chem., 2011, 21, 8975 RSC.
- S. Berger and Y. Drezner, Ferroelectrics, 2005, 321, 85 CrossRef.
- I. Bilecka and M. Niederberger, Nanoscale, 2010, 2(8), 1358 RSC.
- H. Ji, X. Miao, L. Wang, B. Qian and G. Yang, ACS Sustainable Chem. Eng., 2014, 2, 359 CrossRef CAS.
- L. F. da Silva, W. Avansi, M. L. Moreira, A. Mesquita, L. J. Q. Maia, J. Andrés, E. Longo and V. R. Mastelaro, J. Nanomater., 2012, 6 Search PubMed.
- X. H. Zhu and Q. M. Hang, Micron, 2013, 44, 21 CrossRef CAS PubMed.
- S. F. Liu, I. R. Abothu and S. Komarneni, Mater. Lett., 1999, 38, 344 CrossRef CAS.
- A. Cabañas, J. Li, P. Blood, T. Chudoba, W. Lojkowski, M. Poliakoff and E. Lester, J. Supercrit. Fluids, 2007, 40, 284 CrossRef.
- J. Li, G. Mo, Y. Bai and A. Bao, J. Mater. Sci.: Mater. Electron., 2015, 26, 7390 CrossRef CAS.
- M. L. Moreira, G. P. Mambrini, D. P. Volanti, E. R. Leite, M. O. Orlandi, P. S. Pizani, V. R. Mastelaro, C. O. Paiva-Santos, E. Longo and J. A. Varela, Chem. Mater., 2008, 20, 5381 CrossRef CAS.
- S. Das, A. K. Mukhopadhyay, S. Datta and D. Basu, Bull. Mater. Sci., 2008, 31(7), 943 CrossRef CAS.
- V. P. Redko and L. M. Lopato, Inorg. Mater., 1991, 27(9), 1905–1910 CAS.
- J. Wang, Q. Liu and Q. Liu, J. Mater. Chem., 2005, 15, 4141 RSC.
- G. Liu, G. Hong, J. Wang and X. Dong, J. Alloys Compd., 2007, 432(1–2), 200 CrossRef CAS.
- W. Fan, X. Song, Y. Bu, S. Sun and X. Zhao, J. Phys. Chem. B, 2006, 110, 23247 CrossRef CAS PubMed.
- E. van der Kolk and P. Dorenbos, Chem. Mater., 2006, 18(15), 3458 CrossRef CAS.
- E. Mihokova, V. Jary, R. Kucerkova, A. Beitlerova and M. Nikl, DESY Photon Science Annual Report, 2011 Search PubMed.
- W. D. Kingery, H. K. Bowen and D. R. Uhlmann, Introduction to ceramics, A Wiley-Interscience Publication, Canada, 2nd edn, 1976 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra04807b |
| This journal is © The Royal Society of Chemistry 2016 |