
Aerosol-assisted in situ synthesis of iron–carbon composites for the synergistic adsorption and reduction of Cr(vi)
Abstract
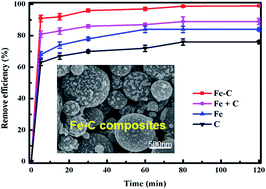
In this work, we adopted newly developed spherical iron–carbon composites to remove hexavalent chromium (Cr(VI)) from contaminated water. These composites were prepared in situ through a facile aerosol-assisted process followed by an economic carbothermal reduction using only common sugar and ferrous sulfate as starting materials, and then characterized by SEM, TEM, XRD and BET. Because of the diverse functions of carbon and nanoscale zero-valent iron, aerosol-assisted iron–carbon composites show synergistic adsorption and reaction towards a more efficient removal of Cr(VI). Under identical experimental conditions, such composites exhibit the highest removal efficiency compared to other materials including nanoscale zero-valent iron particles, aerosol-assisted carbon and their physical mixture. Meanwhile, X-ray photoelectron spectroscopy analysis proved as-prepared iron–carbon composites could effectively transform Cr(VI) to much less toxic trivalent chromium (Cr(III)). These iron–carbon composites produced at low cost show significant potential for the remediation of groundwater and sediment contaminated with heavy metals such as Cr(VI).
 Please wait while we load your content...
Please wait while we load your content...

