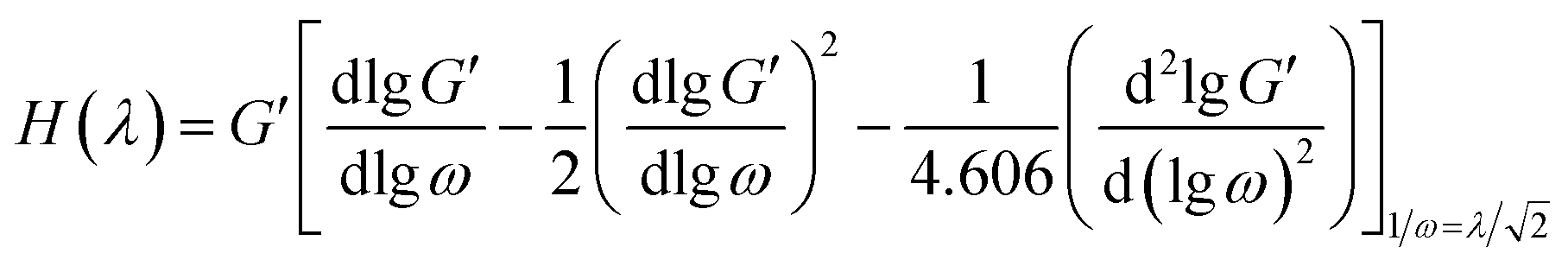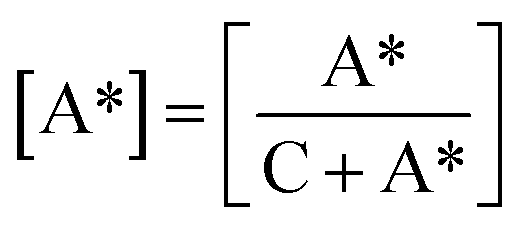DOI:
10.1039/C6RA05133B
(Paper)
RSC Adv., 2016,
6, 62769-62777
Influence of material characteristics on the structure and properties of high-density polyethylene microporous membranes
Received
26th February 2016
, Accepted 15th June 2016
First published on 16th June 2016
Abstract
Five PE resins with different material characteristics were chosen to fabricate microporous membranes based on a melt-stretching mechanism. The lamellar morphology of the initial precursor films and stretched pore structures were characterized. It was found that the precursor films prepared using PE with an appropriate content of larger and small molecular weight species showed higher lamellar thickness and orientation degree. This kind of PE resins exhibited a weak relaxation peak on the left of the main relaxation peak in the relaxation time curves. After stretching, a porosity of around 50% could be obtained and the corresponding air permeability could meet the demands of a separator, which could be used in the field of lithium batteries. Contrary to this, a PE with too many larger molecular weight species was unfavorable for the preparation of microporous membranes, since the higher content of fibre-aligned structures limited the separation of lamellar structures during stretching. PE with too low a content of small molecular weight parts was also inapplicable for the preparation of microporous membranes. The lower lamellar thickness was easy to be deformed and could not support the scaffold of the pore structure, leading to the collapse of the pores and inferior air permeability.
Introduction
As separators, high density polyethylene (HDPE) microporous membranes have been successfully used in the field of Li-ion batteries.1–3 The industrialized method to fabricate this kind of membrane is based on a thermally-induced phase separation mechanism. During the process, a solvent such as dichloromethane has to be used to remove the liquid paraffin to form micropores, leading to some environmental problems. Compared with this, the melt-stretching method using pure PE as the raw material will show no such problem. This kind of method has been used to fabricate polypropylene microporous membranes, which have also been applied in Li-ion batteries as separators.3 During the process, there are three consecutive main stages: (1) production of the precursor film with a row-nucleated lamellar morphology, (2) annealing of the film to thicken the lamellae, and (3) stretching of the film at low temperature to create voids and then stretching at high temperature to enlarge the pores.4 After these stages, the stretched films are heat-set to improve their dimensional stability.
It has been ascertained that the structure of the precursor film has a significant influence on the pore structure and properties of the final stretched microporous membrane. The main factors influencing the precursor row-nucleated lamellar structure include the material’s characteristics and the processing technology, such as the melt-draw ratio, air knife, cast roll and die temperature.5–8 As for the material’s characteristics, usually, high molecular weight resins possessing long chains are good candidates for the generation of a proper row-nucleated lamella since they form long fibrils (threads) that act as sites for lateral lamellae crystallization. The presence of low molecular weight tails (short chains) is generally undesirable for the formation of a row-nucleated structure since it adversely affects the chain relaxation, but it improves melt processability via a smoother extrusion.9 For the preparation of PP microporous membranes, Sadeghi et al. reported that a resin with a higher molecular weight had a tendency to form a planar crystalline morphology as the draw ratio increased.10 In another work, they reported that the addition of up to 10 wt% of a high molecular weight component to a low molecular weight one enhanced the formation of the row-nucleated structure.11 The corresponding membrane surface showed higher pore density and uniform pore size. However, in our previous work, it was found that for PP resins with certain high molecular weight chains, the existence of low molecular weight tails was also important for the formation and stabilization of pore structure.9
For the preparation of PE microporous membranes, Yu12 used two grades of PE with identical number averaged molecular weight, but differing in molecular weight distribution. It was reported that the microporous structure was more uniform for the resin of broader molecular weight distribution. Usually, the melt flow index (MFI) is used to characterize the materials. Some works used PE with an MFI around 0.35 g/10 min to prepare membranes.8,13,14 Contrary to these, Shen et al. compared two kinds of PE with MFI values of 1.08 g/10 min and 5.4 g/10 min, and reported that the PE with a higher MFI was in favour of microvoid formation under extension.15 Tabatabaei et al. used a PE with an MFI of 0.72 g/10 min to prepare a PP/PE/PP trilayer microporous membrane.7
From these works, it seems that PE resins with melt flow indices from 0.35 g/10 min to 5.4 g/10 min can be used to fabricate microporous membranes based on the melt-stretching mechanism. The MFI can only be used to characterize the molecular weight to a certain degree, but could not give detailed material characteristics and behaviour during the practical processing stage. What is the prominent factor of the PE raw materials which determines the structure and properties of a stretched microporous membrane? To solve this problem, in this article, five PE resins with different MFI values were characterized and used to fabricate microporous membranes. The relationship between the material’s characteristics and the properties of the stretched microporous membrane was clarified.
Experimental
Materials
The key characteristics of five PE resins with different MFI values used in this study are presented in Table 1. In this paper, we refer to these materials as PE1, PE2, PE3, PE4 and PE5. PE1 (5021D) was supplied by CNOOC and Shell Petrochemical Company Limited. PE2 (5200B) was from Yanshan petrochemical company, China. PE3 (HY530) was from Japan Polychem Corporation. PE4 (5310E) and PE5 (5000S) were supplied by Yangzi petrochemical company and Lanzhou petrochemical company, respectively. The MFI values were tested under ASTM D1238 conditions of 190 °C and 2.16 kg. To compare their crystalline ability, the non-isothermal half crystallization time was tested by differential scanning calorimetry (DSC; TA Q2000, United States). All the samples were first heated from 60 to 200 °C at a rate of 10 °C min−1 in a nitrogen atmosphere, held at 200 °C for 5 min, then cooled down to 70 °C at 10 °C min−1. The half crystallization time during the non-isothermal crystallization process was calculated. PE2 and PE3 showed approximately the same crystalline ability. Compared with these materials, the crystalline rates of PE4 and PE5 were slower. The molecular weight and polydispersity index were measured using a GPC (Viscotek model 350) at 135 °C and 1,2,4-trichlorobenzene (TCB) as the solvent. The universal calibration curve obtained with polystyrene (PS12000K, APSC) was used to calibrate the corresponding molecular weight of the PE. The GPC curves are shown in Fig. 1 and the corresponding molecular weight values are listed in Table 1. The Mz representing the large molecular weight species of PE1 is much larger than that of the other materials, whereas that of PE5 is smaller. The Mn of PE4 and PE5 is larger than for the other PEs, indicating lesser content of low molecular weight parts.
Table 1 Materials’ characteristics
| |
PE1 |
PE2 |
PE3 |
PE4 |
PE5 |
| Tested under ASTM D 1238 conditions of 190 °C and 2.16 kg. Measured using a GPC (Viscotek model 350) at 135 °C and 1,2,4-trichlorobenzene (TCB) as the solvent. Non-isothermal crystallization time, tested using DSC at a heating and cooling rate of 10 °C min−1. |
| MFI (g/10 min)a |
0.29 |
0.35 |
0.56 |
0.82 |
0.91 |
| Mn |
19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 600 |
20900 |
27900 |
28700 |
26600 |
| Mw |
259000 |
217000 |
206300 |
192000 |
210600 |
| Mz |
1823800 |
1372600 |
1150000 |
1066600 |
946200 |
| PDIb |
13.2 |
10.3 |
7.39 |
6.67 |
7.91 |
| Half crystallization timec |
0.87 |
0.74 |
0.77 |
1.79 |
1.13 |
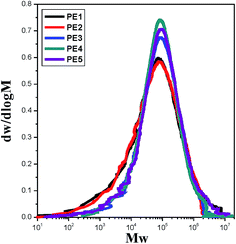 |
| | Fig. 1 GPC curves of PE resins with different melt flow indices. | |
Preparation of precursor film and stretched microporous membrane
The precursor film was prepared by cast extrusion through a T-slot die followed by stretching and thermal setting. During extrusion, the uniaxial (machine direction, MD) stretching was applied to the PE melt, which resulted in oriented crystalline structures. The die temperature was set at 210 °C and a draw ratio of 134 was applied. The draw ratio was determined by the take-up speed, since the extrudate velocity at the exit of the die was constant. The films were produced at a chill roll temperature of 80 °C. The thickness of the precursor film was controlled at 25 μm.
The prepared precursor film was annealed at a taut condition at 125 °C in a hot oven for 2 h. The stretching of the annealed films was carried out using an Instron 5500R machine equipped with a heating chamber. The annealed film was first stretched to 45% at room temperature at a stretching rate of 300 mm min−1 and then stretched to 85% at 120 °C at a stretching rate of 10 mm min−1. After stretching, the film was heat-set at 125 °C for 15 min to improve the dimensional stability.
Characterization
Rheological properties. Dynamic rheological measurements were carried out using an Anton Paar MCR 301 with parallel plate geometry of 25 mm diameter and a gap equal to 1 mm at the temperature of 190 °C. Prior to the frequency sweep tests, time sweep tests at a frequency of 0.628 rad s−1 were performed for two hours to check the thermal stability of the specimens. No degradation (less than 3% change) was observed for the duration of the frequency sweep measurements. The dynamic data were obtained in the linear regime and used to evaluate the weighted relaxation spectrum.
Stress–strain curves and elastic recovery. The stress–strain curves and elastic recovery of the precursor films were tested using an Instron 5500R machine at a deformation rate of 50 mm min−1. Elastic recovery was determined along the stretched direction of the film. The percent elastic recovery (ER%) was calculated using the following equation:| |
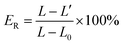 | (1) |
where L0 is the initial length of the film before extension, L is the length when strained to 100% and L′ is the length at the end of extension.
Differential scanning calorimetry (DSC). A TA Q2000 was used to measure the melting curves of the precursor films at a heating rate of 10 °C min−1 under a nitrogen atmosphere. The crystallinity was calculated as:| |
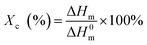 | (2) |
where ΔHm is the measured value of fusion enthalpy and ΔH0m is the fusion enthalpy of a perfectly crystalline PE, that is, 290 J g−1.8
Fourier transform infrared spectroscopy (FTIR). For FTIR spectroscopy, spectra were recorded on a Nicolet 6700 FTIR instrument from Thermo Electron Corp. (DTGS detector, resolution 4/cm, accumulation of 128 scans). The beam was polarized by means of a Spectra-Tech zinc selenide wire grid polarizer from Thermo Electron Corp. The crystalline orientation, based on the difference in the absorption of the plane-polarized radiation, was measured using a vibration in parallel and perpendicular to the stretching machine direction (MD) in the absorbency-FTIR mode. The orientation function was calculated from the following relation.| |
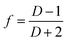 | (3) |
where D is the dichroic ratio and is defined as the ratio of parallel and perpendicular absorbencies of the specific vibration along the melt extrusion direction. For PE, absorption at the wavelength of 1473 cm−1 is due to the contribution of the crystalline phase.
Small-angle X-ray scattering (SAXS). The SAXS measurements were made using an in-house setup with a 30 W micro X-ray source (Incoatec, GmbH), providing a highly parallel beam (divergence about 1 mrad) of monochromatic Cu Kα radiation (λ = 0.154 nm). The scattering intensity was collected using a multiwire proportional chamber detector (Bruker Hi-star) with a resolution of 1024 × 1024 pixels (pixel size of 105 μm). The distance between the sample and detector was 2280 mm.
Wide angle X-ray scattering (WAXS). WAXS was carried out on an X-ray-scattering station with a Mar 345 image plate (3070 × 3070 pixels with a pixel size of 150 μm) as a detector and with a wavelength of 0.154 nm in the National Synchrotron Radiation Laboratory in Hefei (China). The sample-to-detector distance was calibrated to be 299 mm, and the data acquisition time for each frame was 300 s.
Morphology. The surface morphology of the etched precursor films and stretched microporous membranes was characterized using scanning electron microscopy (SEM; LEO SUPRA 55, Carl Zeiss, Germany and S3400, Hitachi, Japan). All the samples were sputtered with a platinum ion beam for 100 s before testing. To clearly observe the crystal arrangement of the cast films, an etching method was used to remove the amorphous part. The films were dissolved in a 0.7% solution of potassium permanganate in a mixture of 35 vol% of orthophosphoric and 65 vol% of sulphuric acid.
Air permeability and porosity. The air permeability of the stretched microporous membranes was characterized using a Gurley Densometer model no. 4150 (Gurley Precision Instruments, New York, USA) according to ASTM D726. The Gurley value was defined as the time required for a specific amount of air (100 mL) to pass through a specific area under a specific pressure (here, 20 kgf cm−2), where a small Gurley value corresponds to high air permeability. The pore size distribution and average pore size were evaluated using a Capillary Flow Porometer (Porous Materials, Inc., New York, USA).3 The porosity was measured using liquid absorption methods according to ASTM D-2873. Paraffin oil was used and the porosity was calculated based on the weight difference before and after immersion in the liquid.
Results and discussion
The rheological properties of five PE resins
To fabricate a microporous membrane based on the melt-stretching mechanism, the first prominent factor is to prepare a precursor film with a row-nucleated crystalline structure, which can be obtained during the extrusion casting, where a high stretching field is applied. Here, the crystalline orientation and morphology strongly depend on the rheological properties of the raw materials. The rheological properties of five PE resins were tested in a frequency range from 0.05 rad s−1 to 300 rad s−1. The storage modulus as a function of frequency is shown in Fig. 2(a). With increasing MFI, the storage modulus shows some decrease.
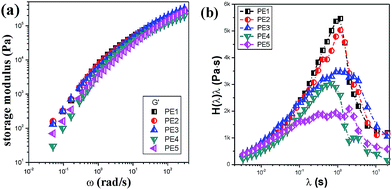 |
| | Fig. 2 Storage modulus (a) and relaxation time spectrum (b) of PE resins with different melt flow indices. | |
To further explain the relaxation behavior of the PE raw materials, the relaxation spectrum (H(λ)) was calculated and is shown in Fig. 2(b); here λ means the relaxation time. H(λ) is a useful method to explain the viscoelastic relaxation of different components. H(λ) can be determined through the Tschoegle approximation16 as revealed in eqn (4):
| |
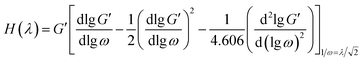 | (4) |
A method suggested by Gramespacher and Meissner17 consists of plotting the relaxation time spectrum H(λ)λ as a function of λ and using the position of the maximum in the resulting curve. According to the equation
where
τd is the relaxation time and
γ is the stretching rate, the Deborah number (
De) could be calculated.
18 Here we assume that the stretching rate distributes evenly within the small stretching distance between the die and cast roll. The stretching rate could be calculated from the difference between the chill roll and the extrudate velocity. In our experiment, during melt extrusion, the melt-draw ratio was set at 134. The calculated
γ is 4.84 s
−1. Therefore, the
De of each PE sample can be calculated. For PE1 and PE2, the main relaxation peak position is approximately the same and the relaxation time is about 1.2 s. The
De is about 5.86, much higher than 1. This means that during melt extrusion, the PE is apt to form an oriented structure. PE3 has a wider relaxation peak than PE1 and PE2, but the intensity of the weighted relaxation peak is a little weaker, meaning the elasticity is reduced due to the small molecular weight. Compared with PE1 and PE2, the corresponding Deborah number of PE3 is still larger than 1. Compared with those of PE1, PE2 and PE3, the relaxation peaks for PE4 and PE5 are wider and weaker. The relaxation time peaks move to the left. The relaxation times of PE4 are located at 0.66 s and 2.82 s, however for PE5, the relaxation times are about 0.33 s, 0.89 s and 1.99 s. The wide relaxation peak indicates a non-uniform chain structure. There should be some more shorter molecular chains in PE5 which induce the appearance of a weak and wide relaxation peak.
The Deborah numbers of PE4 and PE5 are larger than 1, meaning that the PE molecular networks are stretched during the melt-extrusion process and orientation structures are formed.
Structure and properties of precursor films prepared using PE with different melt flow indices
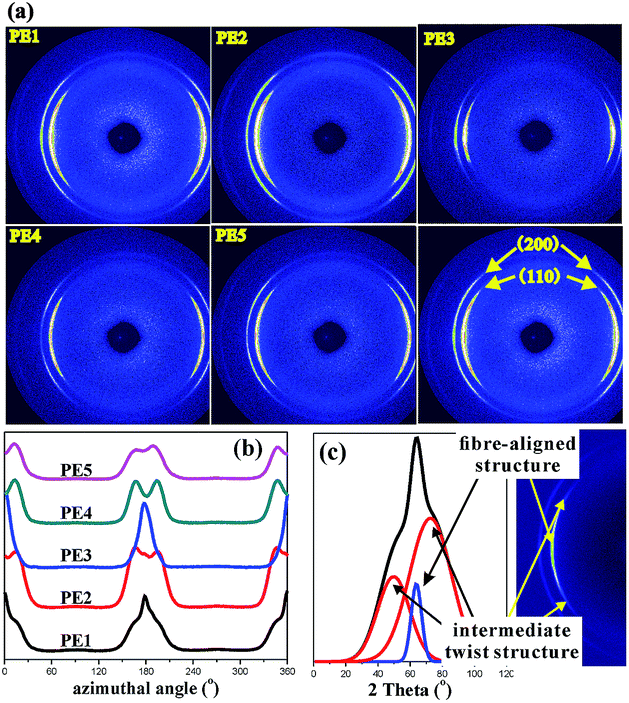
The precursor crystalline structure determines the properties of the stretched microporous membrane. The crystalline morphology, crystalline thickness and orientation degree were characterized. Fig. 3 gives the etched surface morphology of precursor films prepared using PE with different melt flow indices. It can be seen that row-nucleated crystalline structures have been formed for films prepared using PE1, PE2 and PE3. Compared with films prepared with PE1, PE2 and PE3, for the precursor films prepared using PE4 and PE5, the lamellar structure is not uniform and there are some crystalline defects on the surface.
 |
| | Fig. 3 Surface SEM of precursor films prepared using PE with different melt flow indices. | |
Fig. 4 gives the DSC curves of precursor films prepared using PE with different melt flow indices. It is apparent that the melting peak temperature of precursor films prepared using PE2 and PE3 is higher than those of the other films. The lamellar thickness can be calculated by applying the DSC experimental results to the Thomson equation,
| |
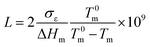 | (6) |
where
Tm is the observed melting point of lamellae with thickness
L,
T0m is the equilibrium melting point of an infinite crystal (
T0m = 415 K for PE),
σε is the surface free energy of the basal plane (
σε = 6.09 × 10
−2 J m
−2 for PE), Δ
Hm is the enthalpy of fusion per unit volume (Δ
Hm = 2.88 × 10
8 J m
−3 for PE).
19 Based on this equation, it can be deduced that the lamellar thicknesses of these two films are larger than those of the other films. For the film prepared using PE1, a weak shoulder appears on the right of the main melting peak. On one hand, this indicates inferior lamellar thickness distribution. On the other hand, as mentioned in the introduction, larger molecular weight species are favorable for the formation of row-nucleated crystalline structures by inducing the formation of long fibrils (threads), which act as sites for lateral lamellae crystallization. The existence of the right shoulder means the existence of long fibrils (shish-structure). The main melting peak comes from the lateral lamellae crystallization. For PE2 and PE3, since the thickness of the lateral lamellae crystallization is higher than that of PE1, the melting peak of lateral lamellae crystallization is combined with that of long fibrils. For PE1, the existence of a much larger molecular weight species induces more formation of long fibrils and less growth in lateral lamellae thickness. Compared with the film prepared with PE2, the melting peak for the film with PE3 is narrow, indicating better lamellar thickness distribution. Based on
eqn (2), the crystallinity was calculated and is listed in
Table 2. The film prepared with PE2 exhibits the largest crystallinity value.
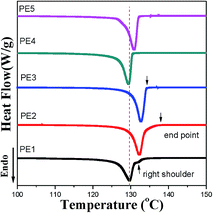 |
| | Fig. 4 Melting curves of precursor films prepared using PE with different melt flow indices. | |
Table 2 Orientation degree, elastic recovery and crystalline properties of precursor films prepared with each sample
| |
PE1 |
PE2 |
PE3 |
PE4 |
PE5 |
| Orientation degree |
0.204 |
0.266 |
0.264 |
0.173 |
0.171 |
| Elastic recovery (%) |
43.8 |
57.8 |
46.7 |
42.6 |
43.9 |
| Long period (nm) |
23.6 |
26.6 |
25.7 |
22.3 |
22.9 |
| Crystalline thickness (nm) |
15.0 |
17.3 |
17.0 |
14.3 |
14.6 |
| Amorphous region thickness (nm) |
8.6 |
9.3 |
8.7 |
7.8 |
8.3 |
| FWHM |
0.139 |
0.150 |
0.143 |
0.115 |
0.105 |
| Xc (%) |
78.0 |
87.7 |
81.1 |
64.8 |
84.7 |
To further investigate the lamellar structure of precursor films prepared using PE with different melt flow indices, 2D-WAXS and 2D-SAXS were used. In the WAXS patterns shown in Fig. 5, two lattice planes representing the α-crystals of PE from the inner to outer circles indexed as (110) and (200) are clearly visible. All the samples show symmetrical arc diffraction patterns in an equatorial direction. This means that highly-oriented crystals are obtained during the melt-stretching process. In an effort to clearly elucidate the lamellar structure of the main lamellar crystals, azimuthal scans of the (110) lattice plane as a function of azimuthal angle are displayed in Fig. 5(b). The diffraction peak of the (110) lattice plane can be separated into two parts. The pair of 2-point equatorial peaks located at 0° and 180° are related to the fibre-aligned structure crystals, and another four point pattern corresponds to a system of intermediate twisting lamellae,20 as shown in Fig. 5(c). It is clear that different PE raw materials show non-uniform diffraction peaks relating to different relaxation behavior and stretching induced crystallization properties, which demonstrates the different content of fibre-aligned structure crystals and twisting lamellae crystals. To determine the fibre-aligned structure content within the different MFI PEs, the area percentage of fibre-aligned structures was calculated using Peakfit software. The relative content was calculated based on:
| |
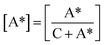 | (7) |
where C represents intermediate twisting lamellae and A* represents fibre-aligned structure crystals in
Fig. 6(c) after subtraction of the baseline. The relative content of fibre-aligned structure crystals is shown in
Fig. 6.
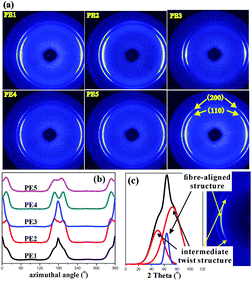 |
| | Fig. 5 2D-WAXS images for PE cast films with different MFI (a), the azimuthal-integrated intensity of the 110 crystallographic plane for different samples (b), and the schematic of peak separation (c). | |
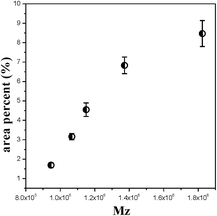 |
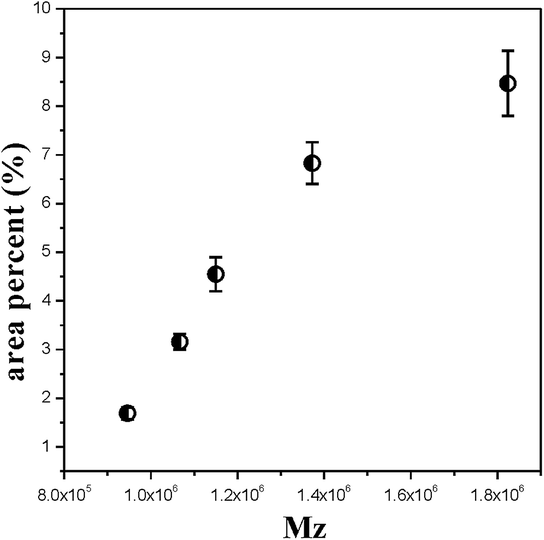
| | Fig. 6 The area percentage of fibre-aligned structure crystals for PE cast films with increasing Mz. | |
Fig. 6 shows that with increasing Mz, the content of fibre-aligned structure crystals increases under the same extrusion conditions. This result is similar to that of the shish-kebab crystal formation process. The shish crystal or the fibre-aligned crystal will be formed when the molecular weight is higher than the critical molecular weight during the stretching process.21 With the content of higher molecular weight parts increasing, the long chains tend to form an extended fibre-aligned structure. However, the short chains tend to form a twisting lamellae crystal. It is obvious that the long molecular chains have more influence on fibre-aligned crystal formation.
Fig. 7 gives the 2D-SAXS pattern and one-dimensional scattering intensity distribution along the machine direction, as well as the corresponding correlation function. The scattering intensity was integrated along the meridional direction using sector integration. Before integration, the instrument background was subtracted considering sample absorption. In the one-dimensional scattering intensity distribution, no multiplication of q2 to I(q) is performed because of the anisotropic orientation of the lamellae in the samples,22 where q is the scattering vector,
| | |
q = 4πsinθ/λ
| (8) |
λ is the X-ray wavelength, 0.154 nm, and 2
θ is the scattering angle. It is apparent that the melt stretching induces a pronounced meridional pattern along the
qy direction, indicating the formation of an oriented crystalline structure, in agreement with the above SEM results. The one-dimensional correlation function,
K(
r), is a powerful tool in revealing the morphological parameters of the crystalline-amorphous two-phase systems and is obtained from indirect Fourier transformation of the scattering function. As indicated by the insert image in
Fig. 7(b), the curve gives various structural parameters,
e.g. the crystalline long period (
L) and amorphous region thickness (
La), according to the well-known methodology proposed by Strobl
et al.23 It must be mentioned that it is impossible to decide whether it is the amorphous or the crystalline thickness that is read out from the correlation function without prior knowledge of the crystallinity. In the present study, the degree of crystallinity as shown in
Table 2 is higher than 50%, ensuring the assignment of the smaller value obtained from the correlation function to the average thickness of the amorphous region. The crystalline thickness (
Lc) can be obtained with the relation,
in which
L is the long period. In addition, the scattering patterns for films prepared with different melt flow indices show different widths and distances to the beam. The width of the SAXS patterns is contributed to by the orientation of the crystalline lamellae and the lateral size of the lamellae.
24 Generally, the narrow full width at half maximum (FWHM) corresponds to a highly oriented crystalline structure
25 or long lateral lamellae dimension.
26 The
L,
Lc,
La and FWHM for precursor films with different melt flow indices are also included in
Table 2. The films prepared using PE2 and PE3 show larger long periods and crystalline thicknesses. The latter crystalline thickness information is in agreement with the above-mentioned higher melting peak in the DSC curves. PE2 and PE3 exhibit fast crystalline ability. During the melt extrusion process, there is enough time for them to crystallize, leading to higher lamellar thickness. The higher orientation degree should induce narrow scattering patterns and lower FWHM values. But the results in
Table 2 are contrary to this. The larger FWHM values for films prepared using PE2 and PE3 indicate that for these two films, the lateral lamellar size is shorter.
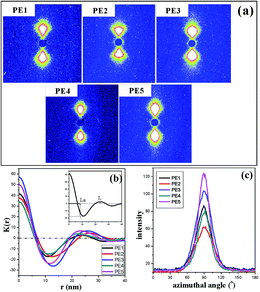 |
| | Fig. 7 SAXS pattern (a), corresponding one-dimensional scattering intensity distribution (b), and the azimuthal-integrated intensity of the SAXS pattern for PE with different melt flow indices (c). | |
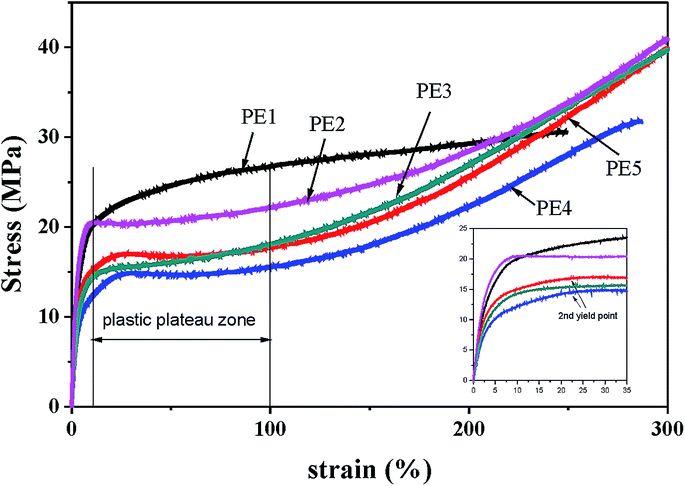
The crystalline orientation degree calculated from the FTIR results and the elastic recovery are also listed in Table 2. The elastic recovery can be used as an indicator for characterizing the elastic deformation properties of lamellar crystalline structures. It can be seen that the precursor films prepared using PE2 and PE3 show higher crystalline orientation degree and elastic recovery. PE4 and PE5 exhibit shorter relaxation times. During melt extrusion, they are easy to disorient, leading to lower orientation degree. For the films prepared using PE2 and PE3, the higher orientation degree and larger crystalline thickness result in their higher elastic recovery. For the film prepared using PE1, although its orientation degree is higher than those of PE4 and PE5, the thin crystalline thickness and non-uniform lamellar structure lead to a decrease in elastic recovery. Fig. 8 gives the stress–strain curves of precursor films prepared using PE with different melt flow indices. It is surprising to find that with an increase in the melt-flow index, three kinds of stress–strain curve appear. For PE1, after the yield point, with an increase in strain, the stress is increasing. This may be related to its larger Mz value and formation of more long fibrils. For PE2 and PE3, after the yield point, a plastic shoulder appears before the strain-hardening region. Sadeghi et al.6 summarized that the following phenomena took place during stretching at room temperature: void formation as a result of short tie chain scission, crystal blocks slipping and reorienting along the stretching direction, stretching of longer tie chains as shorter tie chains were broken apart, and crystallization of highly stretched long tie chains, leading to the strain-hardening phenomenon. In our previous work,27 it has been proved that the existence of a plastic shoulder is related to the stretching of tie chains and to the formation of initial pores during stretching. It can be seen that the formation of pore structure is highly related to the content of tie chains. The volume fractions of tie chains were calculated according to the method proposed by Takayanagi,28
| |
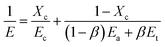 | (10) |
where
Xc is the crystallinity,
Ec,
Ea and
Et are the modulus of the crystalline part, amorphous region and tie chains, respectively,
β is the volume fraction of tie chains, and
E is the elastic modulus calculated from the initial part before the yield point in the stress–strain curves. During stretching, the contribution of the amorphous region to the modulus can be neglected and then the volume fraction of tie chains can be calculated based on
| |
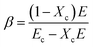 | (11) |
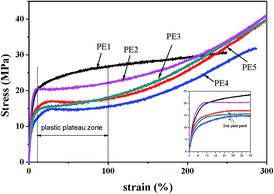 |
| | Fig. 8 Stress–strain curves of precursor films prepared using PE with different melt flow indices. | |
Here, the modulus of the tie chains is supposed to be approximately that of the crystalline part and 300 GPa is used.29 The calculated fraction of tie chains is listed in Table 3. PE1 exhibits the highest content of tie chains, which could be attributed to its higher Mz.
Table 3 Tie content of precursor films prepared using PE with different melt flow indices
| |
E (MPa) |
Tie content (%) |
| PE1 |
1523.5 |
0.112 |
| PE2 |
1433.7 |
0.059 |
| PE3 |
1284.4 |
0.081 |
| PE4 |
779.1 |
0.049 |
| PE5 |
960.7 |
0.057 |
Compared with PE2 and PE3, PE4 shows the lowest tie chain fraction. For the film prepared using PE1, a higher tie chains fraction leads to difficulty in breakage of the short tie chains to form the plastic shoulder. During stretching, higher stress is needed. For PE4 and PE5, after the yield point, a second yield point appears. This is related to the non-uniform lamellar structure in these films. For these two films, the orientation degree is lower than that of the other films.
The structure and properties of stretched microporous membranes
Fig. 9 shows the morphology of stretched microporous membranes prepared using PE with different melt flow indices. It is apparent that for the membranes prepared using PE2 and PE3, pronounced pores and bridges connecting the separated lamellae appear. For PE1, PE4 and PE5, there are some areas on the surface with no pores. In sample PE1, the high content of fibre-aligned structure limits the lamellae crystals’ separation to form pore structures. The lamellae crystals appear to show shearing deformation accompanying the destroyed fibre-form. Contrary to this, the lamellae are directly stretched to be broken. Many pore closed areas and cracks are shown on the surfaces of PE4 and PE5. The crystallinity, porosity and Gurley values of these microporous membranes are listed in Table 4. It can be seen that compared with the annealed film, the crystallinity of the microporous membranes is greatly decreased after stretching. During stretching, some crystalline parts are destroyed. Compared with Fig. 3 and 9, it can be clearly seen that the lamellae show typical slip and deformation after stretching. For the membranes prepared using PE2 and PE3, the porosity is higher than 45%, whereas for the other samples the porosity is lower than 25%. This is in agreement with the above SEM results. The membranes prepared using PE2 and PE3 show lower Gurley values, indicating better air permeability properties. This kind of air permeability property is acceptable for application in the field of lithium batteries as a separator.
 |
| | Fig. 9 SEM of stretched microporous membranes prepared using PE with different melt flow indices. | |
Table 4 Porosity and Gurley values of microporous membranes prepared using PE with different melt flow indices
| |
Crystallinity (%) |
Gurley value (s/100 mL) |
Porosity (%) |
| PE1 |
57.3 |
9392 |
11.4 |
| PE2 |
64.1 |
221 |
48.8 |
| PE3 |
63.0 |
192 |
50.7 |
| PE4 |
42.5 |
12014 |
3.7 |
| PE5 |
53.5 |
3923 |
24.4 |
Relationship between material characteristics and properties of stretched microporous membranes
The above results show that a better pore structure is formed in the membranes prepared using PE2 and PE3 and they show better air permeability properties. Contrary to this, inferior air permeability appears in the membranes prepared using PE1, PE4 and PE5.
For PE2 and PE3, both larger molecular weight tail and smaller molecular weight parts exist, exhibiting a higher relaxation time of around 1.2 s and a lower relaxation time of around 0.4 s. The larger molecular weight tail affords the formation of long fibrils during extrusion casting. The smaller molecular weight parts support the crystalline ability, showing higher crystalline rates, resulting in larger lamellar thickness, although the lamellar lateral size is smaller. Higher orientation degree and lamellar thickness result in better lamellae arrangement. After stretching, the separation of uniform lamellar structures leads to higher porosity and better air permeability.
For PE1, the Mz representing a higher molecular weight species is too large. Although there are some small molecular weight parts, the higher molecular weight species are a prominent factor in controlling the lamellar morphology. During extrusion casting, more long fibrils are formed, leading to the existence of the right shoulder in the DSC curve. The corresponding lamellar thickness is small and the whole orientation degree is low. In addition, the higher molecular weight species leads to the formation of a higher content of tie chains. During stretching, no plastic region appears and after the yield point with increasing strain, the stress increases, indicating higher stress is needed to separate the lamellar structure. This will lead to a decrease in pores.
For PE4, the lower content of small molecular weight parts and the crystalline rate are not beneficial for stress-induced crystallization, leading to smaller lamellar thickness and a lower orientation degree. For PE5, the higher molecular weight species are not enough for the formation of fibre-aligned structures, resulting in lower orientation. The lower crystalline rate is also unfavorable for the growth of a lamellar structure. The non-apparent relaxation peak indicates a non-uniform molecular structure. All these induce the lower orientation degree and lamellar thickness. During stretching, the second yield point appears and the thin lamellae cannot support the formed pore structure, resulting in the deformation of the lamellar structure and collapse of the formed pores. The direct result is the disappearance of pores. The possible pore formation process related to different PE resins is shown in Fig. 10.
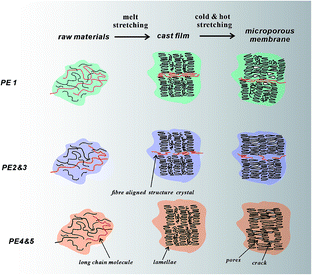 |
| | Fig. 10 Schematic of initial lamellar structure and stretched pores for PE with different material characteristics. | |
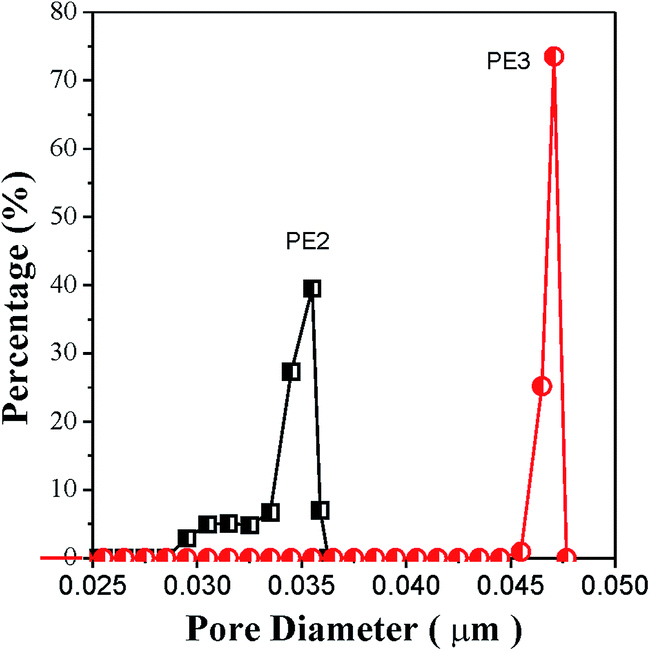
Compared with the membrane prepared using PE2, the porosity of the membrane prepared using PE3 is 1.9% larger and the corresponding Gurley value is 29 s lower. Although the precursor films prepared using PE2 and PE3 show approximately the same crystalline orientation degree and lamellar thickness, the film prepared using PE3 shows a larger tie chains fraction. It has been reported that the formation of pores is related to the stretching of tie chains during room-temperature and hot stretching. The stretching of tie chains with a larger fraction results in larger pore size. The direct result is higher porosity and a lower Gurley value. Fig. 11 gives the pore size distribution of microporous membranes prepared using PE2 and PE3. It is apparent that the membrane prepared using PE3 shows a larger pore size.
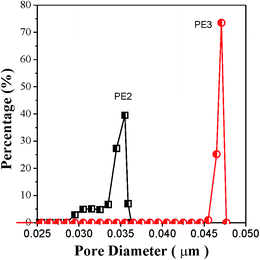 |
| | Fig. 11 Pore size distribution of microporous membranes prepared using PE2 and PE3. | |
Conclusions
In this article, microporous membranes were prepared using five PE resins with different material characteristics. The orientation degree, lamellar thickness and tie chain content of the precursor films were tested. For PE1 with a larger Mz value, the tie chains content is 0.112%, the orientation degree is 0.204 and the crystalline thickness is 15 nm. For PE2 and PE3, with a certain content of small molecular weight parts and larger molecular weight species, the tie chains content is about 0.06–0.08%, the orientation degree is about 0.26 and the crystalline thickness is around 17 nm. For PE4 and PE5 with a lower content of small molecular weight parts, the tie chains content is about 0.05–0.06%, the orientation degree is only 0.17 and the crystalline thickness is only 14.5 nm. For PE1, the existence of a larger Mz is not beneficial for the lamellar separation during stretching, whereas for PE4 and PE5, the thinner lamellae thickness could not support the pore scaffold. Only microporous membranes prepared using PE2 and PE3 show porosity of up to 48% and better air permeability, showing their application ability as separators in the lithium battery field.
Acknowledgements
The authors would like to thank the National Science Foundation of China under Grant No. 51003017, the Project of High Level Talents in Higher School and the Guangdong Provincial Major Key Projects of Applied Research, Development of Science and Technology (2015B090925021) and the Natural Science Foundation of Guangdong Province under Grant No. 2016A030310344 for financial support. They also want to thank Shenzhen Senior Materials Company, Ltd., for generously supplying raw materials.
Notes and references
- S. S. Zhang, J. Power Sources, 2007, 164, 351 CrossRef CAS.
- T. Tagawa and K. Ogura, J. Polym. Sci., Polym. Phys. Ed., 1980, 18, 971 CrossRef CAS.
- R. J. Xu, X. D. Chen, J. Y. Xie, Q. Cai and C. H. Lei, Ind. Eng. Chem. Res., 2015, 54, 2991 CrossRef CAS.
- F. Sadeghi, Developing of microporous polypropylene by stretching. PhD thesis, Ecole Polytechnique de Montreal, Canada, 2007.
- F. Sadeghi, A. Ajji and P. J. Carreau, J. Membr. Sci., 2007, 292, 62 CrossRef CAS.
- F. Sadeghi, A. Ajji and P. J. Carreau, J. Plast. Film Sheeting, 2005, 21, 199 CrossRef CAS.
- S. H. Tabatabaei, P. J. Carreau and A. Ajji, J. Membr. Sci., 2009, 345, 148 CrossRef CAS.
- S. Y. Lee, S. Park and S. Song, Polymer, 2006, 47, 3540 CrossRef CAS.
- C. H. Lei, S. Q. Wu, R. J. Xu, X. L. Peng, W. Q. Shi and B. Hu, Polym. Eng. Sci., 2013, 53, 2594 Search PubMed.
- F. Sadeghi, A. Ajji and P. J. Carreau, Polym. Eng. Sci., 2007, 47, 1170 Search PubMed.
- F. Sadeghi, A. Ajji and P. J. Carreau, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 148 CrossRef CAS.
- T. H. Yu and G. L. Wilkes, Polymer, 1996, 37, 4675 CrossRef CAS.
- J. Kim, S. S. Kim, M. Park and M. Jang, J. Membr. Sci., 2008, 318, 201 CrossRef CAS.
- J. Liu, Z. T. Ding, Z. Y. Liu and M. B. Yang, Acta Polym. Sin., 2011, 54, 1278 CrossRef.
- L. Q. Shen, Z. K. Xu and Y. Y. Xu, J. Appl. Polym. Sci., 2002, 84, 203 CrossRef CAS.
- N. W. Tschoegle, The Phenomenological Theory of Linear Viscoelastic Behavior, Spinger-Verlag, Berlin, 1989 Search PubMed.
- H. Gramespacher and J. J. Meissner, J. Rheol., 1992, 36, 1127 CrossRef CAS.
- S. Coppola, N. Grizzuti and P. L. Maffettone, Macromolecules, 2001, 34, 5030 CrossRef CAS.
- Y. An, J. J. Holt, G. R. Mitchell and A. Vaughan, Polymer, 2006, 47, 5643 CrossRef CAS.
- K. H. Bader, Advances in Physics Theories and Applications, 2015, 41, 13 Search PubMed.
- R. H. Somani, B. S. Hsiao, A. Nogales, H. Fruitwala, S. Srinivas and A. H. Tsou, Macromolecules, 2000, 33, 9385 CrossRef CAS.
- L. Fu, Z. Jiang, H. F. Enderle, D. Lilge, Z. Wu, S. S. Funari and Y. F. Men, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 716 CrossRef CAS.
- G. Strobl, The physics of polymers, Spinger, Berlin, 2nd edn, 1997 Search PubMed.
- Y. Tang, Z. Jiang, Y. Men, L. An, H. F. Enderle, D. Lilge and J. Rieger, Polymer, 2007, 48, 5125 CrossRef CAS.
- A. Prasada, R. Shroff, S. Rane and G. Beaucag, Polymer, 2001, 42, 3103 CrossRef.
- R. J. Xu, X. D. Chen, Q. Cai, C. B. Chen, Y. F. Lin, C. H. Lei and L. B. Li, RSC Adv., 2015, 5, 27722 RSC.
- C. H. Lei, W. L. Huang, R. J. Xu and Y. Q. Xu, J. Plast. Film Sheeting, 2012, 28, 151 CrossRef CAS.
- M. Takayanagi and K. Nitta, Macromol. Theory Simul., 1997, 6, 181 CrossRef CAS.
- R. Seguela, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 1729 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.