
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phosphine-free chiral iridium catalysts for asymmetric catalytic hydrogenation of simple ketones†
Prashant
Kumar
*ab,
Torsten
Irrgang
b,
George E.
Kostakis
 a and
Rhett
Kempe
*b
a and
Rhett
Kempe
*b
aDepartment of Chemistry, School of Life Sciences, University of Sussex, Brighton BN1 9QJ, UK. E-mail: pbhukumar@gmail.com
bInorganic Chemistry II, University of Bayreuth, 95440-Bayreuth, Germany. E-mail: kempe@uni-bayreuth.de
First published on 5th April 2016
Abstract
Novel pyridylalkylamine and aminopyridinato ligand stabilized iridium complexes with no P ligand are introduced. These complexes have been investigated as catalysts for asymmetric hydrogenation of simple ketones, resulting in an active catalyst for bulky alkyl aryl ketones that is α-methylpropiophenone. The ligands were synthesized from inexpensive starting materials and their modular design allows for the introduction of a broad variety of substitution patterns. Additionally, better activity and selectivity was observed at 20 °C and 20 bar H2 pressure with a catalyst loading as low as 0.05 mol% iridium. These phosphorus free catalysts have always been a central issue in both academic and industrial research.
1. Introduction
The catalytic reduction of polar multiple bonds-mainly carbonyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) functionalities into corresponding hydroxyl (–CH–OH) functionalities by molecular hydrogen has great significance in modern synthetic chemistry. The stereoselective hydrogenation of ketones to yield enantiomerically pure alcohols is a key step, in the synthesis of fine chemicals, perfumes, and pharmaceuticals. This reaction is normally performed by complexes of precious metals (e.g. Ru, Rh, Ir) using either H2 or iPrOH as the hydrogen source.1 In particular, the reduction of ketones with gaseous hydrogen provides an atom-economical synthetic method. High enantioselectivity, low catalyst loadings, quantitative yields, atom economy and mild conditions are attractive features of this transformation as evident in the ever growing list of research articles that use these methods. The importance of this topic was recognized when Knowles2,3 and Noyori3,4 were awarded the 2001 Nobel prize for their instrumental contributions to this field. The development of highly enantioselective chemocatalyst based on their low toxicity and lower price ligands could drastically increase the importance of asymmetric homogeneous catalysis.5 Therefore, it has been well established that organometallic complexes of ruthenium have been well-employed in this scenario, recently in particular by the groups of Noyori6 and Braunstein.7 Iridium and rhodium complexes have long been known to be highly effective homogeneous hydrogenation catalysts, with RhCl(PPh3)3 (Wilkinson's catalyst)8 and the most widely used [Ir(cod)(py)(PCy3)]PF6 [py = pyridine, cod = 1,5-cyclooctadiene, Crabtree's catalyst9]. In contrast, iridium phosphorus-free catalysts are extensively less efficient and/or enantioselective in this asymmetric hydrogenation.10 Relatively, few organometallic complexes derived from amine based ligands have been reported to be effective at the control of asymmetric hydrogenation reactions,10,11 particularly in comparison to the large numbers of diphosphine12,13 and mixed amine/phosphine14,15 ligands, which have been reported. In principle, amine-based ligands possess a potential advantage over phosphorus because they are relatively simple to prepare and less prone to decomposition and oxidation reactions. Furthermore, iridium(I) complexes of cycloocta-1,5-diene (cod) are of interest because the coordinated cod can be replaced by other ligands. Cod can also be hydrogenated to provide vacant coordination sites around iridium and thus increase the catalytic activities of the complexes.16 In this paper, we report the synthesis and structure of phosphorus-free iridium catalysts containing aminopyridinato and pyridylalkylamine (Scheme 1) and cod ligands for the hydrogenation of simple ketones.
O) functionalities into corresponding hydroxyl (–CH–OH) functionalities by molecular hydrogen has great significance in modern synthetic chemistry. The stereoselective hydrogenation of ketones to yield enantiomerically pure alcohols is a key step, in the synthesis of fine chemicals, perfumes, and pharmaceuticals. This reaction is normally performed by complexes of precious metals (e.g. Ru, Rh, Ir) using either H2 or iPrOH as the hydrogen source.1 In particular, the reduction of ketones with gaseous hydrogen provides an atom-economical synthetic method. High enantioselectivity, low catalyst loadings, quantitative yields, atom economy and mild conditions are attractive features of this transformation as evident in the ever growing list of research articles that use these methods. The importance of this topic was recognized when Knowles2,3 and Noyori3,4 were awarded the 2001 Nobel prize for their instrumental contributions to this field. The development of highly enantioselective chemocatalyst based on their low toxicity and lower price ligands could drastically increase the importance of asymmetric homogeneous catalysis.5 Therefore, it has been well established that organometallic complexes of ruthenium have been well-employed in this scenario, recently in particular by the groups of Noyori6 and Braunstein.7 Iridium and rhodium complexes have long been known to be highly effective homogeneous hydrogenation catalysts, with RhCl(PPh3)3 (Wilkinson's catalyst)8 and the most widely used [Ir(cod)(py)(PCy3)]PF6 [py = pyridine, cod = 1,5-cyclooctadiene, Crabtree's catalyst9]. In contrast, iridium phosphorus-free catalysts are extensively less efficient and/or enantioselective in this asymmetric hydrogenation.10 Relatively, few organometallic complexes derived from amine based ligands have been reported to be effective at the control of asymmetric hydrogenation reactions,10,11 particularly in comparison to the large numbers of diphosphine12,13 and mixed amine/phosphine14,15 ligands, which have been reported. In principle, amine-based ligands possess a potential advantage over phosphorus because they are relatively simple to prepare and less prone to decomposition and oxidation reactions. Furthermore, iridium(I) complexes of cycloocta-1,5-diene (cod) are of interest because the coordinated cod can be replaced by other ligands. Cod can also be hydrogenated to provide vacant coordination sites around iridium and thus increase the catalytic activities of the complexes.16 In this paper, we report the synthesis and structure of phosphorus-free iridium catalysts containing aminopyridinato and pyridylalkylamine (Scheme 1) and cod ligands for the hydrogenation of simple ketones.
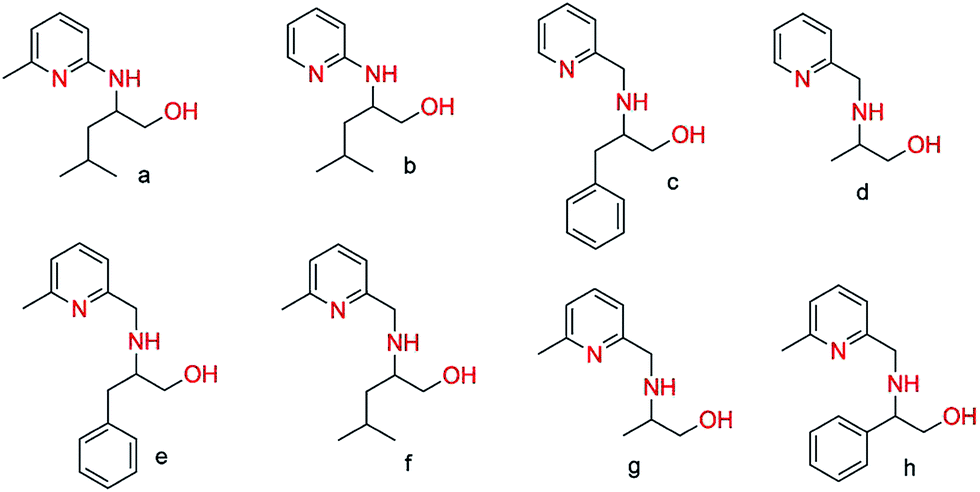 | ||
| Scheme 1 Aminopyridinato (a, b) and pyridylalkylamine (c–h) ligands. | ||
2. Experiment section
2.1. General method
All reactions and manipulations which are sensitive to moisture or air were performed in argon-filled glove box or using standard Schlenk techniques. Non-halogenated solvents were distilled from sodium benzophenone ketyl and halogenated solvents from P2O5. Deuterated solvents were obtained from Cambridge Isotope Laboratories and were degassed, dried and distilled prior to use. [Ir(OMe)(cod)]2 (cod = 1,5-cyclooctydiene, Me = methyl) was prepared according to the literature.17 All chemicals were purchased from commercial vendors. 1H and 13C NMR spectra were obtained at room temperature on ARX 250 or Varian INNOVA 300 or 400 spectrometer using C6D6, and CDCl3 as a solvent and chemical shifts are reported in ppm relative to the deuterated solvent. Elemental analyses for C, H and N were performed on a Vario elementary EL III apparatus.2.2. Synthetic procedures
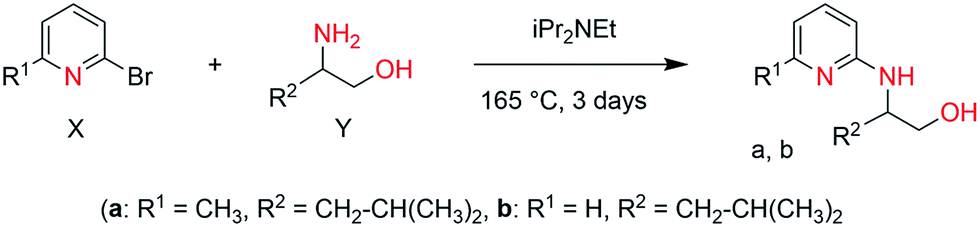
General procedure. A 25 mL medium-pressure tube charged with 2-bromopyridine (1.00 mmol), or 6-methyl-2-bromopyridine (1.00 mmol) X, substituted amino alcohol (1.00 mmol) Y, N,N-diisopropyl ethylamine (1.16 mmol) and a stir bar was flushed with nitrogen several times, stoppered and heated at 160 ± 5 °C for 2 days. The tube was allowed to cool to room temperature, the contents was diluted with a small amount of CH2Cl2 and chromatographed (hexanes–EtOAc 2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 → 1:1 → neat EtOAc) to afford the product (a, b) in good yield.
:1 → 1:1 → neat EtOAc) to afford the product (a, b) in good yield.
4-Methyl-2-(6-methyl-pyridin-2-yl-amino)-pentan-1-ol (a). Yellow viscous liquid, 64% yield. C12H20N2O (208.16): calcd C 69.19, H 9.68, N 13.45; found C 68.73, H 10.01, N 12.97. 1H NMR (250 MHz, CDCl3, 298 K): δ = 7.26 (t, J = 3.6 Hz, 1H, Ar); 6.37 (d, J = 5.2 Hz, 1H, Ar); 6.25 (d, J = 4.5 Hz, 1H, Ar); 3.78–3.70 (m, 2H, CH2–OH); 3.49–3.45 (m, 1H, CH–NH); 2.29 (s, 3H, CH3), 1.69–1.64 (m, 1H, CH–(CH3)2); 1.45–1.30 (m, 2H, CH2–CH–(CH3)2); 0.88–0.83 (dd, J = 5.2 Hz, 6H, CH–(CH3)2) ppm. 13C NMR (62.90 MHz, CDCl3, 298 K): δ = 158.14 (C5, pyridine), 155.81 (C1, pyridine), 137.96 (C3, pyridine), 111.93 (C4, pyridine), 104.79 (C2, pyridine), 67.65 (CH2, CH2OH), 52.97 (CH, CHNH); 41.06 (CH2, CH2CH(CH3)2); 24.87 (CH–(CH3)2); 23.69, 22.93 (CH–(CH3)2); 22.22 (CH3) ppm.
4-Methyl-2-(pyridin-2-yl-amino)-pentan-1-ol (b). White solid, 40% yield. C11H18N2O (194.14): calcd C 68.01, H 9.34, N 14.42; found C 67.86, H 9.21, N 14.41. 1H NMR (250 MHz, CDCl3, 298 K): δ = 7.99 (d, J = 5.4 Hz, 1H, Ar); 6.95 (t, J = 7.4 Hz, 1H, Ar); 6.23 (t, J = 5.5 Hz, 1H, Ar); 5.94 (d, J = 7.4 Hz, 1H, Ar); 3.66–3.61 (m, 2H, CH2–OH); 3.48–3.47 (m, 1H, CH–NH); 1.48–1.46 (m, 1H, CH–(CH3)2); 1.14–1.08 (m, 2H, CH2–CH–(CH3)2); 0.78–0.74 (dd, J = 5.2 Hz, 6H, CH–(CH3)2) ppm. 13C NMR (62.90 MHz, CDCl3, 298 K): δ = 158.94 (C5, pyridine); 147.88 (C1, pyridine); 137.03 (C3, pyridine); 112.71 (C2, pyridine); 108.38 (C4, pyridine); 52.23 (CH2, CH2OH); 44.99 (CH, CHNH); 41.07 (CH2, CH2CH(CH3)2); 24.99 (CH–(CH3)2); 22.18, 23.05 (CH–(CH3)2) ppm.
General procedure. To ice-cooled solution of amino alcohol (1.00 mmol) (Y), pyridine-2-carbaldehyde or 6-methylpyridine-2-carbaldehyde (1.00 mmol) (X′) was added slowly in 20 mL of methanol. The resulting solution was stirred at room temperature for 3 h and subsequently treated with sodiumtetrahydridoborate (2.5 mmol) in small portions. After additional 2 h, water (50 mL) was added and the reaction mixture was concentrated to about 10 mL using rotovap. The remaining solution was extracted with dichloromethane (2 × 20 mL), and the organic layer was separated and subsequently dried with sodium sulphate and concentrated in vacuum to afford the product (c–h) in good to high yield.
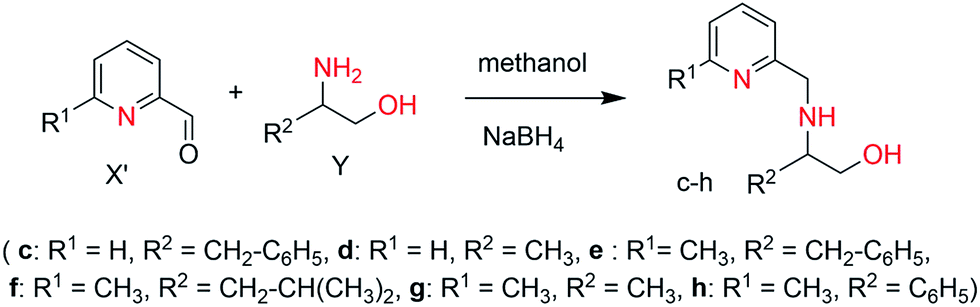
3-Phenyl-2-[(pyridin-2-yl-methyl)-amino]-propan-1-ol (c). Yellow solid, 90% yield. mp 65 °C. C15H18N2O (242.14): calcd C 73.35, H 7.49, N 11.56; found C 74.43, H 7.31, N 11.32. 1H NMR (250 MHz, CDCl3, 298 K): δ = 8.46 (d, J = 1.4 Hz, 1H, Ar), 7.56 (t, J = 3.8 Hz, 1H, Ar), 7.24–7.08 (m, 5H, Ar), 3.97–3.82 (m, 3H, CH2–N, CH–N), 3.63–3.58 (m, 2H, CH2–O), 2.73–2.95 (m, 2H, CH2-Ar) ppm. 13C NMR (62.90 MHz, CDCl3, 298 K): δ = 160.11 (C5, pyridine), 157.85 (C1, pyridine), 139.68 (C9, phenyl), 136.42 (C3, pyridine), 129.65 (C10, phenyl), 128.49 (C11, phenyl), 126.21 (C12, phenyl), 121.11 (C4, pyridine), 118.96 (C2, pyridine), 62.61 (CH2, CH2OH), 60.97 (CH, CHNH), 52.31 (CH2, CH2NH), 38.66 (CH2, CH2C6H5) ppm.
2-[(Pyridin-2-yl-methyl)-amino]-propan-1-ol (d). Yellow solid, 90% yield. mp 70 °C. C9H14N2O (166.11): calcd C 65.03, H 8.49, N 16.85; found C 64.89, H 8.38, N 16.27. 1H NMR (250 MHz, CDCl3, 298 K): δ = 8.38 (d, J = 1.8 Hz, 1H); 7.06 (t, J = 5.2, 1H); 6.91 (d, J = 3.6, 1H); 6.60 (t, J = 6.5 Hz, 1H); 3.85–3.70 (m, 2H, CH2–N); 3.55–3.52 (m, 2H, CH2–O); 2.70–2.66 (m, 1H, CH–NH); 0.92 (d, J = 3.2, 3H, CH3). 13C NMR (62.90 MHz, CDCl3, 298 K): δ = 160.53 (C5, pyridine); 149.176 (C1, pyridine); 136.00 (C3, pyridine); 122.11 (C4, pyridine); 121.67 (C2, pyridine); 65.63 (CH2, CH2OH); 54.70 (CH, CHNH); 52.33 (CH2, CH2NH); 17.16 (CH3, CH3CH) ppm.
2-[(6-Methyl-pyridin-2-yl-methyl)-amino]-3-phenyl-propan-1-ol (e). Yellow solid, 91% yield. mp 76 °C. C16H20N2O (256.16): calcd C 73.86, H 7.86, N 10.93; found C 73.96, H 7.69, N 10.68. 1H NMR (250 MHz, CDCl3, 298 K): δ = 7.44 (t, J = 1.4 Hz, 1H, Ar), 7.29–7.16 (m, 5H, Ar), 6.92 (t, J = 3.8 Hz, 1H, Ar), 3.91–3.82 (m, 3H, CH2–N, CH–N), 3.60–3.41 (m, 2H, CH2–O), 2.91–2.79 (m, 2H, CH2-Ar), 2.48 (s, 3H, CH3) ppm. 13C NMR (62.90 MHz, CDCl3, 298 K): δ = 157.84 (C5, pyridine), 158.83 (C1, pyridine), 138.88 (C9, phenyl), 136.93 (C3, pyridine), 129.29 (C10, phenyl), 128.43 (C11, phenyl), 126.24 (C12, phenyl), 121.63 (C4, pyridine), 119.13 (C2, pyridine), 62.57 (CH2, CH2OH), 60.75 (CH, CHNH), 51.93 (CH2, CH2NH), 38.24 (CH2, CH2C6H5), 24.19 (CH3) ppm.
4-Methyl-2-[(6-methyl-pyridin-2-yl-methyl)-amino]-propan-1-ol (f). Pale yellow solid, 94% yield. mp 60 °C. C13H22N2O (222.17): calcd C 70.23, H 9.97, N 12.60; found C 70.44, H 10.20, N 12.51. 1H NMR (250 MHz, CDCl3, 298 K): δ = 7.23 (d, J = 2.4 Hz, 1H, Ar), 6.63 (d, J = 4.3 Hz, 1H, Ar), 3.88–3.83 (d, J = 5.4, 2H, CH2–NH); 3.83–3.74 (dd, 1H, CH–NH); 3.49–3.43 (dd, 1H, CH2–OH); 2.80–2.49 (m, 1H, CH2–OH); 2.72 (s, 3H, CH3), 1.78–1.67 (sept, 1H, CH–(CH3)2); 1.45–1.30 (m, 2H, CH2–CH–(CH3)2); 0.86–0.82 (t, J = 6.4 Hz, 6H, CH–(CH3)2) ppm. 13C NMR (62.90 MHz, CDCl3, 298 K): δ = 160.20 (C5, pyridine), 157.91 (C1, pyridine), 136.59 (C3, pyridine), 121.26 (C4, pyridine), 119.32 (C2, pyridine), 63.62 (CH2, CH2OH), 57.35 (CH, CHNH), 52.21 (CH2, CH2NH); 41.61 (CH2, CH2CH(CH3)2); 25.06 (CH–(CH3)2); 24.20, 23.15 (CH–(CH3)2); 22.91 (CH3) ppm.
2-[(6-Methyl-pyridin-2-yl-methyl)-amino]-propan-1-ol (g). Brown solid, 84% yield. mp 75 °C. C10H16N2O (180.13): calcd C 66.63, H 8.95, N 15.54; found C 66.38, H 8.64, N 15.09. 1H NMR (250 MHz, CDCl3, 298 K): δ = 7.04 (t, J = 2.6 Hz, 1H); 6.79 (d, J = 3.6, 1H); 6.55 (d, J = 4.5 Hz, 1H); 3.85–3.70 (m, 2H, CH2–N); 3.37–3.35 (m, 2H, CH2–O); 2.71–2.69 (m, 1H, CH–NH); 2.35 (s, 3H, CH3); 0.94 (dd, 3H, CH3) ppm. 13C NMR (62.90 MHz, CDCl3, 298 K): δ = 159.91 (C5, pyridine); 157.98 (C1, pyridine); 136.45 (C3, pyridine); 121.14 (C4, pyridine); 119.39 (C2, pyridine); 65.49 (CH2, CH2OH); 55.02 (CH, CHNH); 52.50 (CH2, CH2NH); 23.99 (CH3); 17.21 (CH3, CH3CH) ppm.
2-[(6-Methyl-pyridin-2-yl-methyl)-amino]-2-phenyl-ethanol (h). Yellow solid, 84% yield. mp 60 °C. C15H18N2O (242.14): calcd C 74.35, H 7.49, N 11.56; found C 74.23, H 7.07, N 11.56. 1H NMR (250 MHz, CDCl3, 298 K): δ = 7.44 (t, J = 1.4 Hz, 1H, Ar), 7.29–7.16 (m, 5H, Ar), 6.92 (t, J = 3.8 Hz, 1H, Ar), 3.91–3.82 (m, 3H, CH2–N, CH–N), 3.60–3.41 (m, 2H, CH2–O), 2.91–2.79 (m, 2H, CH2-Ar), 2.48 (s, 3H, CH3) ppm. 13C NMR (62.90 MHz, CDCl3, 298 K): δ = 159.72 (C5, pyridine); 157.98 (C1, pyridine); 141.69 (C9, phenyl); 136.45 (C3, pyridine); 129.29 (C10, phenyl); 128.89 (C11, phenyl); 126.38 (C12, phenyl); 121.33 (C4, pyridine); 119.39 (C2, pyridine); 67.62 (CH2, CH2OH); 65.10 (CH, CHNH); 52.89 (CH2, CH2NH); 23.97 (CH3) ppm.
4-Methyl-2-(6-methyl-pyridin-2-yl-amino)-pent-1-oxy-iridium(I)-cyclooctadiene (1a). [{IrOMe(cod)}2] 0.662 g (1.00 mmol) was dissolved in THF (20 mL) and a solution of 0.416 g (2.00 mmol) aminopyridinato ligands (a) dissolved in THF (5 mL) was subsequently added dropwise. A red solution was obtained after addition. The reaction solution was stirred overnight at room temperature. The resulting solution was removed in vacuo and the residue was recrystallised from toluene, yielding red crystals at −30 °C, which were suitable for X-ray crystal structure analyses. Yield: 0.52 g (65%). C28H42Ir2N2O (807.04): calcd C = 41.67; H = 5.25; N = 3.47; found C = 41.92; H = 5.16; N = 3.86. 1H NMR (250 MHz, C6D6, 296 K): δ = 7.43 (t, 1H); 6.34 (dd, 2H, Ar); 4.45–4.36 (m, 4H, COD); 3.80–3.38 (m, 6H, CH2–OH, COD); 2.66–2.58 (m, 1H, CH–NH); 2.54–2.27 (m, 6H, CH–CH2–CH, COD); 2.07 (s, 3H, CH3), 1.75–1.39 (m, 5H, CH–(CH3)2/CH2, COD); 0.96–0.78 (dd, 6H, CH–(CH3)2) ppm. 13C (250 MHz, C6D6): 158.29, 157.02, 139.0, 114.96, 106.69 (C5H3N); 71.47 (CH2, CH2OH), 68.71, 64.63, 62.41, 60.28, 60.03 (COD); 53.95 (CH, CHNH); 41.77 (CH2, CH2CH(CH3)2); 41.56, 35.72, 34.07, 33.94, 32.82, 32.25, 32.06, 31.96, 30.57, 30.28, 25.42 (COD); 24.88 (CH–(CH3)2); 23.60, 23.42, (CH–(CH3)2); 22.52 (CH3) ppm.
4-Methyl-2-(pyridin-2-yl-amino)-pent-1-oxy-iridium(I)-cyclooctadiene (1b). Complex 1b was prepared using 0.388 g (2.00 mmol) aminopyridinato ligand (b), following the above procedure for 1a. Red crystals were obtained in hexane at room temperature. Yield: 0.50 g (63%). C27H40Ir2N2O (793.01): calcd C = 40.89; H = 5.08; N = 3.53; found C = 41.12; H = 4.98; N = 3.90. 1H NMR (250 MHz, C6D6, 296 K): δ = 7.06 (t, 1H); 6.27 (dd, 2H, Ar); 4.50–4.34 (m, 4H, COD); 3.84–3.46 (m, 6H, CH2–OH, COD); 2.77–2.70 (m, 1H, CH–NH); 2.57–2.26 (m, 6H, CH–CH2–CH, COD); 1.49–1.25 (m, 5H, CH–(CH3)2, COD); 0.81–0.71 (dd, 6H, CH–(CH3)2) ppm. 13C (250 MHz, C6D6): 159.23, 148.12, 139.79, 115.96, 108.40 (C5H4N); 76.16 (CH2, CH2OH), 66.42, 66.12, 64.86, 64.56, 55.33 (COD); 51.17 (CH, CHNH); 44.99 (CH2, CH2CH(CH3)2); 42.09, 41.24, 35.58, 34.34, 33.96, 32.41, 30.99, 28.09, 27.20, 25.19 (COD); 24.55 (CH–(CH3)2); 23.17, 22.63, (CH–(CH3)2) ppm.
3-Phenyl-2-[(pyridin-2-yl-methyl)-amino]-prop-1-oxy-iridium(I)-cyclooctadiene (2c). To an yellow solution of 0.484 g (2.00 mmol) pyridylalkylamine ligand (c) in 20 mL of THF, 0.662 g (1.00 mmol) [{IrOMe(cod)}2] dimer was added, accompanied by a color change to dark red. The reaction solution was stirred at room temperature for 16 h. The resulting solution was filtered and the filtrate concentrated to dryness under vacuum. The remaining solid was washed with 15 mL hexane and subsequently re-crystallized from toluene. Yellow crystals were grown at −30 °C, which were suitable for X-ray crystal structure analyses. Yield: 0.64 g (68%). C38H48Ir2N2O (933.18): calcd C = 48.91; H = 5.18; N = 3.00; found C = 48.45; H = 5.26; N = 2.86. 1H NMR (250 MHz, C6D6, 296 K): δ = 7.90 (d, 1H); 7.75 (d, 1H); 7.68 (d, 1H); 7.52 (d, 1H); 7.33–6.99 (m, 5H, C6H5); 5.49 (br, 1H, NH); 4.55–4.40 (m, 4H, COD); 3.60–3.36 (m, 4H, COD); 3.12 (s, 2H, CH2NH); 3.00 (d, CH, CH2O); 2.69–2.62 (m, CH, CHNH); 2.27–2.16 (m, 10H, CH2C6H5, COD); 1.64–1.48 (m, 8H, COD) ppm. 13C (250 MHz, C6D6): δ = 165.16, 147.14, 135.79, 134.99, 129.59 (C5H3N); 129.34, 126.30, 124.78, 122.50 (C6H5); 68.08 (CH, –CH2O); 67.18 (CH, CHN); 66.24, 65.16, 61.06, 59.86 (COD); 55.0 (CH2, CH2NH); 40.73, 35.67, 33.05, 32.14, 31.68, 31.29, 31.03, 30.74, 30.70, 28.14, 28.74, 22.76 (COD) ppm.
2-[(Pyridin-2-yl-methyl)-amino]-prop-1-oxy-iridium(I)-cyclooctadiene (2d). Complex 2d was prepared using 0.332 g (2.00 mmol) pyridylalkylamine ligand (d), following the above procedure for 2c. At −30 °C red crystals were obtained. Yield: 0.49 g (57%). C32H44Ir2N2O (857.09): calcd C = 44.84; H = 5.17; N = 3.27; found C = 44.80; H = 4.96; N = 3.01. 1H NMR (250 MHz, C6D6, 296 K): δ = 7.82 (d, 1H); 7.55 (d, 1H); 7.27 (d, 1H); 7.25 (d, 1H); 5.25 (br, 1H, NH); 4.42–4.29 (m, 4H, COD); 4.20–4.14 (m, 4H, COD); 3.94–3.90 (m, 2H, CH2NH); 3.40 (d, CH, CH2O); 2.91–2.88 (m, CH, CHNH); 2.05–1.93 (m, 8H, COD); 1.54–1.34 (m, 8H, COD); 0.89 (d, 3H, CH3) ppm. 13C (250 MHz, C6D6): δ = 165.56, 147.39, 140.72, 124.91, 122.62 (C5H4N); 76.91, 68.02, 67.57 (COD); 66.82 (CH2, CH2OH); 54.34 (CH, CHNH); 51.33 (CH2, CH2NH); 49.0, 31.62, 30.87, 30.70, 30.55, 30.12, 29.82, 28.55, 28.14, 28.02, 25.71, 23.21, 22.70 (COD); 16.16 (CH3, CH3CH) ppm.
2-[(6-Methyl-pyridin-2-yl-methyl)-amino]-3-phenyl-prop-1-oxy-iridium(I)-cyclooctadiene (2e). Complex 2e was prepared using 0.512 g (2.00 mmol) pyridylalkylamine ligand (e) following the above procedure for 2c. Yellow crystals were grown at room temperature, which were suitable for X-ray crystal structure analyses. Yield: 0.38 g (69%). C24H31IrN2O (555.71): calcd C = 51.86; H = 6.12; N = 5.13; found C = 51.96; H = 6.16; N = 5.17. 1H NMR (250 MHz, C6D6, 296 K): δ = 7.34 (d, 1H); 7.27 (d, 1H); 7.23 (d, 1H); 7.20–6.35 (m, 5H, C6H5); 4.16–4.15 (m, 1H, COD); 4.09–4.04 (m, 2H, COD); 3.91–3.90 (m, 1H, COD); 3.88 (s, 2H, CH2NH); 3.61 (d, CH, CH2O); 3.52–3.46 (m, CH, CHNH); 2.94 (d, CH, CH2C6H5); 2.52 (s, 3H, CH3), 2.39–2.27 (m, 4H, COD); 2.23–1.89 (m, 4H, COD) ppm. 13C (250 MHz, C6D6): δ = 158.8, 155.95, 138.79, 136.57, 130.00 (C5H3N); 129.28, 126.91, 122.37, 118.50 (C6H5); 74.2 (CH, –CH2O); 70.5 (CH, CHN); 61.38, 58.03 (CH, COD); 55.9 (CH2, CH2NH); 52.07, 51.7 (CH, COD); 38.9 (CH, CH2–C6H5); 33.85, 32.44, 31.35, 31.14 (CH2, COD); 25.5 (CH3) ppm.
Complexes 2f–h: not isolated, pre-catalyst-solution prepared via alcohol elimination in situ.
3. Results and discussion
3.1. Ligand synthesis
The targeted ligands were obtained from commercially available reactants in one or two steps based on previous studies.21 The ligand precursors 4-methyl-2-(6-methyl-pyridin-2-yl-amino)-pentan-1-ol (a) and 4-methyl-2-(pyridin-2-yl-amino)-pentan-1-ol (b) were synthesized in a one-pot reaction starting from 6-methyl 2-bromopyridines and 2-bromopyridines with enantiopure amino alcohols using N,N-diisopropylethyl amine as a base at 165 °C for 3 days (Scheme 2) in quantitative yields.22 Ligands (c–h) were prepared by reductive amination of heteroaromatic aldehydes (pyridine-2-carbaldehyde or 6-methylpyridine-2-carbaldehyde) with enantiopure aminoalcohols over two steps including the formation of the corresponding ketimine and further reduction with sodium borohydride (Scheme 3). Both methodologies allowed us to prepare a family of substituted ligands based on a pyridylalkylamine and aminopyridinato core. All the synthesized ligands are air stable and soluble in polar as well as non-polar solvents and characterized by 1H, 13C NMR and elemental analysis.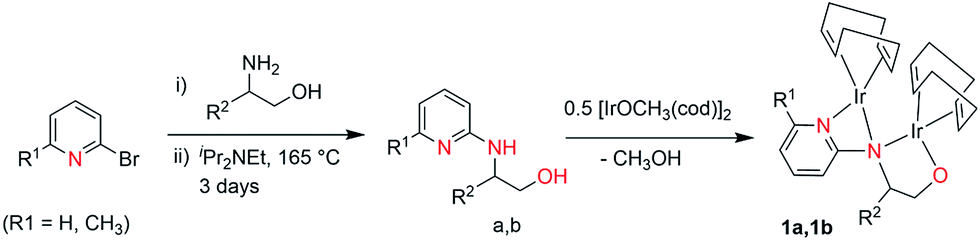 | ||
| Scheme 2 Synthesis of iridium complexes 1 (a) R1 = CH3, R2 = CH2–(CH3)2, (b) R1 = H, R2 = CH2–CH(CH3)2. | ||
 | ||
| Scheme 3 Synthesis of iridium complexes 2 (c) R1 = H, R2 = CH2–C6H5, (d) R1 = H, R2 = CH3, (e) R1 = CH3, R2 = CH2–C6H5, (f) R1 = CH3, R2 = CH2–CH(CH3)2, (g) R1 = CH3, R2 = CH3, (h) R1 = CH3, R2 = C6H5. | ||
3.2. Synthesis and structure of the complexes
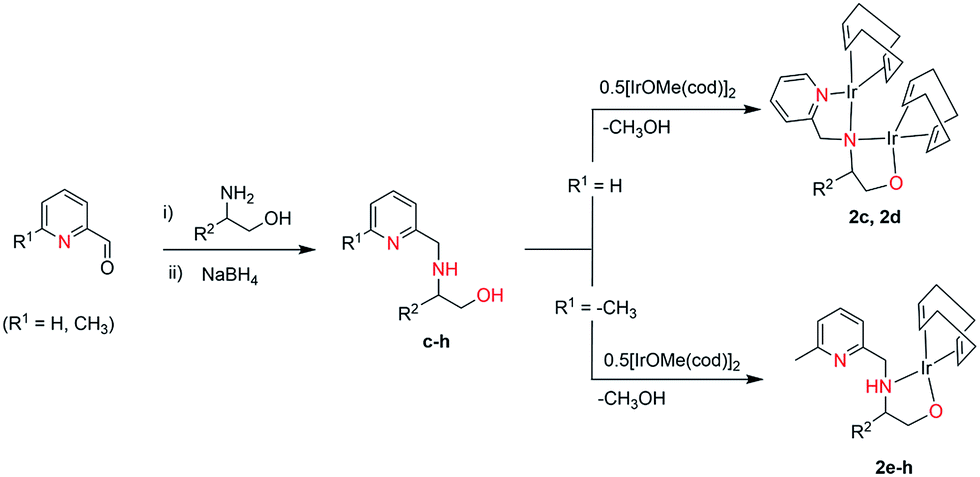
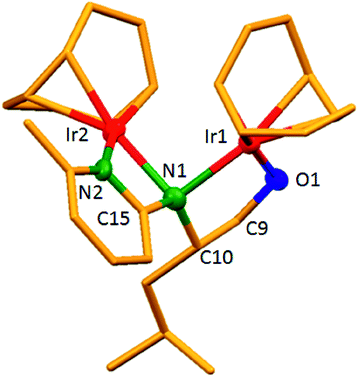
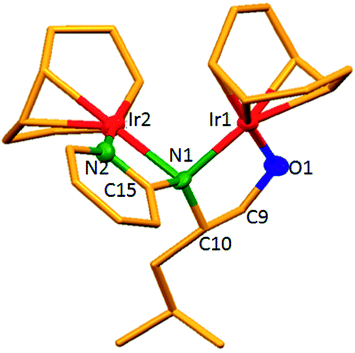
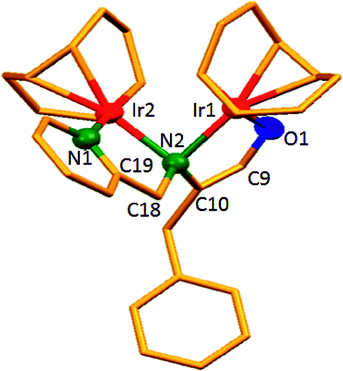
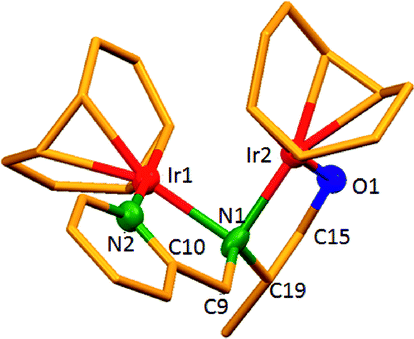
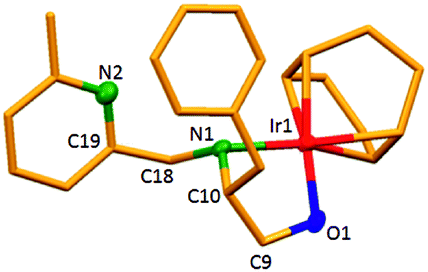
The dinuclear iridium complexes 1a, b and 2c, d can be obtained via alcohol elimination route with [IrOCH3(cod)]2 (ref. 23) (Scheme 2 and 3). Addition of 0.5 equiv. of the metal precursor to a solution of a–d in THF gives rise to 1a, b and 2c, d which is accompanied by a color change to yellow/reddish-brown. While under the same reaction condition mononuclear iridium complexes 2e–h can be synthesized by reaction with [IrOCH3(cod)]2 (Scheme 3). This synthetic approach shows the presence of the methyl group, at sixth position of pyridine ring, and the methylene (–CH2NH) group, favour the formation of mononuclear iridium complex.X-ray crystal structure analyses of the dinuclear complexes (1a, b and 2c, d) and the mononuclear (2e) were performed to determine the molecular structures (Fig. 1–5). Selected crystallographic data and geometrical parameters are summarized in the Fig. 1–5, respectively and details about the data collection, solution, and refinement are summarized in the ESI (Table S1†). In complexes 1a and 1b (Fig. 1 and 2) the Ir1-atoms are stabilized by the formation of a five membered chelate (Ir1, N1, C9, C10, O1) and Ir2-atom is stabilized by the formation of a four membered chelate (Ir2, N1, N2, C15). It was observed that Ir–Namido bond [Ir2–N1 2.145(10) 1a, Ir2–N1 2.202(5), Å] is significantly longer than Ir–Npyridine bond [Ir2–N2 2.020(10) 1a, N2–Ir2 2.062(6), Å 1b]. These simply suggest that the anionic charge of the ligand is not localized at the amido-N-atom. The Ir–O bond is significantly shorter than the Ir–N bond indicating the localization of the negative charge of the alkoxy (–CH2OH) at the oxygen atom. The bite angle N1–Ir1–O1 (80.6°) is almost equal to 2c, but significantly smaller than 2e. While in 2c and 2d, the two iridium atoms (Ir1 and Ir2) are stabilized by the formation of a five membered chelate (Ir1, N2, C9, C10, O1, and Ir2, N1, N2, C19, C18 2c; Ir2, N1, C15, C16, O1, and Ir1, N1, N2, C9, C10 2d). Again, the Ir–O bond [2.021(6) Å] is significantly shorter than the Ir–N bond [2.151(6) Å] and Ir–N bond [2.103(7) Å] and Ir–N bond [2.106(6) Å] are almost equal in 2c and 2d (Fig. 3 and 4).
 | ||
| Fig. 1 Molecular structure of 1a. [Thermal ellipsoid diagram (50%) for all non-carbon atoms] without H atoms. Selected bond length [Å] and angles [°]: Ir1–O1 1.979(8), Ir1–N1 2.144(10), Ir2–N1 2.145(10), Ir2–N2 2.020(10), O1–C9 1.413(15), N1–C10 1.490(15), N1–C15 1.388(15); N1–Ir1–O1 80.6(4), N1–Ir2–N2 63.5(4), Ir2–N1–Ir1 94.0(4), Ir1–O1–C9 114.9(7), C10–N1–Ir2 130.7(7), C10–N1–Ir1 106.0(7). | ||
 | ||
| Fig. 2 Molecular structure of 1b. [Thermal ellipsoid diagram for all non-carbon atoms] without H atoms. Selected bond length [Å] and angles [°]: Ir1–O1 1.998(5), Ir1–N1 2.086(6), Ir2–N1 2.202(5), Ir2–N2 2.062(6), O1–C9 1.401(9), N1–C10 1.491(9), N1–C15 1.414(11); N1–Ir1–O1 80.3(2), N1–Ir2–N2 63.5(2), Ir2–N1–Ir1 82.73(16), Ir1–O1–C9 115.2(5), C10–N1–Ir2 108.1(14), 128.4(4), C10–N1–Ir1 109.9(4). | ||
 | ||
| Fig. 3 Molecular structure of 2c. [Thermal ellipsoid diagram for all non-carbon atoms] without H atoms. Selected bond length [Å] and angles [°]: Ir1–O1 2.021(6), Ir1–N2 2.151(6), Ir2–N1 2.103(7), Ir2–N2 2.106(6), O1–C9 1.356(9), N2–C10 1.452(10); N2–Ir1–O1 80.9(2), N1–Ir2–N2 79.7(2), Ir2–N2–Ir1 89.7(2), Ir1–O1–C9 112.4(5), C10–N2–Ir2 119.0(5), C18–N2–Ir1 119.1(5), C18–N2–Ir2 109.8(5), C10–N2–Ir1 107.1(5), N2–C18–C19 112.2(6). | ||
 | ||
| Fig. 4 Molecular structure of 2d. [Thermal ellipsoid diagram for all non-carbon atoms] without H atoms. Selected bond length [Å] and angles [°]: Ir2–O1 2.017(12), Ir1–N1 2.145(14), Ir2–N1 2.150(18), Ir1–N2 2.089(16), O1–C16 1.39(2), N1–C15 1.47(3); N1–Ir2–O1 80.6(6), N1–Ir1–N2 80.6(7), Ir2–N1–Ir1 88.7(6), Ir2–O1–C16 114.4(11), C15–N1–Ir2 108.1(14), C15–N1–Ir1 125.6(13), C9–N1–Ir1 109.6(11), C9–N1–Ir2 118.3(13), C10–N2–Ir1 116.1(13), N1–C9–C10 112.3(15). | ||
 | ||
| Fig. 5 Molecular structure of 2e. [Thermal ellipsoid diagram for all non-carbon atoms] without H atoms. Selected bond length [Å] and angles [°]: Ir1–O1 1.995(3), Ir1–N1 2.127(3), O1–C9 1.425(5), C9–C10 1.530(6), N1–C18 1.492(6); N1–Ir1–O1 82.67(13), C9–O1–Ir1 112.7(3), C9–C10–N1 106.1(4), C10–N1–Ir1 105.9(3), N1–C18–C19 110.4(4). | ||
Furthermore, in complexes 2e (Fig. 5) the monoanionic ligand coordinates the Ir-atom via a five membered chelate (Ir1, N1, C9, C10, and O1). The Ir1–O1 bond [1.995(3) Å] is significantly shorter than the Ir1–N1 bond [2.127(3) Å], simply indicating the localization of the anionic charge of the alkoxy at the oxygen atom. The 6-methylpyridyl moiety is bent out of the pyridine plane (N1–C18–C19 110.4°) and coordination by the pyridine nitrogen does not occur. However, the bite angle of complex 1a (N1–Ir1–O1, 80.6°), 1b (N1–Ir1–O1, 80.3), 2c (N2–Ir1–O1, 80.9°), and 2d (N1–Ir1–O1, 80.6°) are almost same but slightly smaller than 2e (N1–Ir1–O1, 82.67°). The increased angle brought the Ir atom, the reaction center of the catalyst, closer to the ligand backbone and created a chiral environment around the Ir atom that permitted more efficient catalysis.
3.3. Catalytic studies
Based on the pioneering work of Kempe et al. group24 where amido iridium complexes were employed as a catalyst for the asymmetric hydrogenation of the simple ketones, we have chosen to determine the catalytic potential of iridium–N^N^O complexes for such reactions. In general, the screening reactions were performed using 2.29 mmol of the substrate at 20 °C for 48 h and the catalyst was prepared in situ from stock solutions of [IrOCH3(cod)]2 and N^N^O ligand.First of all, mononuclear iridium complexes 2e–h (Scheme 3) were used to determine the effect of substitution at the amino skeleton of the ligands on the asymmetric hydrogenation of simple ketones (Table 3). Evidently, the catalysts 2e and 2f containing benzyl and isopropyl substituents on the nitrogen (Table 1, entries 2 and 4) gave better results than those containing the methyl (2g) or phenyl (2h) substituent (Table 1, entries 6 and 8). The best catalyst for this reaction seems to be compound 2e (Table 1, entry 2), which achieved a >99% conversion and 89% ee at a very low catalyst loading (0.05 mol% iridium) and excess KOtBu.
In this catalytic work, all optimization studies were carried out with α-methyl propiophenone as model substrates. As can be seen from Table 2, KOtBu appears to be the most suitable base, because complete conversion and a good ee (89%) could only be achieved by using this base (Table 2, entry 6). This result shows that KOtBu is needed as an additive for better enantioselectivity during hydrogenation. Presence of base KOtBu is essential for catalysis, because base itself act as a catalyst for the hydrogenation of simple ketones under drastic conditions,25 its potassium cations accelerate the reaction rate of phosphane–ruthenium–diamine complexes.26
:1, 48 h, 0.05 mol% 2e, 20 °C, and H2 (20 bar)
| Entry | Base | Conv. [%] | ee [%] |
|---|---|---|---|
| 1 | — | 55 | 10 |
| 2 | NaNH2 | 25 | 65 |
| 3 | KOSiMe3 | 40 | 34 |
| 4 | KOH | 64 | 30 |
| 5 | NaOtBu | 46 | 74 |
| 6 | KOtBu | >99 | 89 |
| 7 | KN(SiMe3)2 | 69 | 80 |
After these optimisations, we examined the addition of excess amounts of base was needed to allow complete conversion or whether catalytic quantities of base are sufficient. Therefore, the influence of the base/catalyst ratio was investigated (Table 3, entries 1–5). The results shown in Table 3, entries 1–5, suggest that it is necessary to use a base/catalyst ratio of 500:1, because only in this case (Table 3, entry 5) was it possible to obtain complete conversion and a better ee (96%) within 48 h in the presence of catalytic amount of acetone (acetone:α-methylpropiophenone 2:1).
:α-methyl-propiophenone 2:1, THF, 48 h, 0.05 mol% 2e, 20 °C, 20 bar
| Entry | Base:pre-catalyst |
Conv. (%) | ee (%) |
|---|---|---|---|
| 1 | 1:1 (0.05 mol%) |
60 | 32 |
| 2 | 10:1 (0.5 mol%) |
78 | 60 |
| 3 | 50:1 (2.5 mol%) |
96 | 82 |
| 4 | 100:1 (5 mol%) |
>99 | 90 |
| 5 | 500:1 (25 mol%) |
>99 | 96 |
However, at a base loading of only 5 mol%, it was possible to achieve a conversion (>99%) and ee (90%) (Table 3, entry 4), which contradicts the aforementioned excess requirement for base. For this reason, we investigated whether it is possible to bring the reaction to complete conversion or better ee by the use of 25 mol% of KOtBu. To this end, the reaction time was 48 h an ee of 96% were obtained.
The final screening was performed on the catalyst loading to find the minimum catalyst loading necessary to achieve full conversion and good ee (Fig. 6). As shown in Fig. 6, it was sufficient to use a catalyst loading of 0.05 mol% to obtain a very good ee (96%) for this reaction (Fig. 6). It was observed the addition of KOtBu and acetone leads to quantitative conversion with 2e as a pre-catalyst with catalyst loadings of 1, 0.5, 0.2, 0.1, and 0.05 mol% (Fig. 6). It was examined that without KOtBu and acetone with 1.0 mol% of 2e a conversion of 55% and 10% ee is obtained (Fig. 6, Table 1, and entry 1). The catalytic performance for hydrogenation of α-methylpropiophenone of 2e–h with KOtBu and acetone was examined (Table 4). Upon addition of acetone in the presence of KOtBu, 96% ee could be achieved under same reaction condition with 0.05 mol% catalyst loading (Table 4, entry 2). The increase in enantioselectivity could result from the fact that the more enantioselective catalyst species is formed during catalysis, the addition of (non-prochiral) ketones, which is hydrogenated in parallel and preferentially faster, should support the formation of a more enantioselective catalyst.
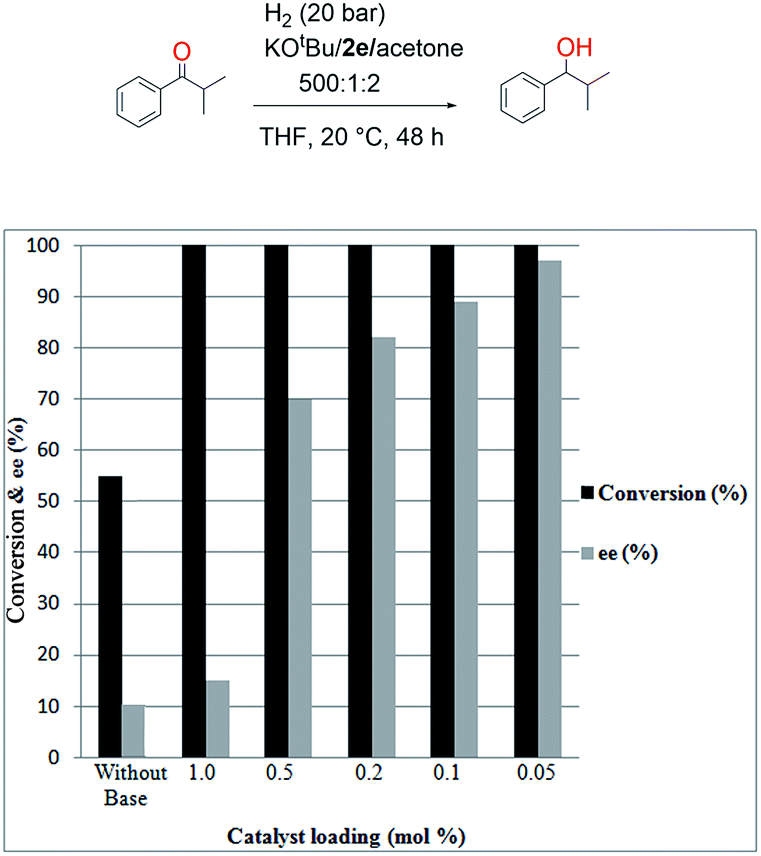 | ||
| Fig. 6 Hydrogenation of α-methylpropiophenone with 2e in the presence of KOtBu and acetone. | ||
As it can be seen in Table 4, catalyst 2e and 2f are active catalyst for asymmetric hydrogenation of α-methylpropiophenone. Further, we were interested to examine the effect of acetone on the reaction. It was observed that acetone/pre-catalyst 2e ratio of 2:1 was use to obtain complete conversion (>99%) and good ee (96%). It was also observed that if we changed the acetone/catalyst ratio (1:1 and 3:1), considerably affect on the conversion (86% and 90%) and ee (80% and 86%).
To confirm the results we achieved for the hydrogenation α-methylpropiophenone with catalyst 2e, different simple aromatic ketones were further examined (Fig. 7). These results show that the reduction of the hindered aromatic ketones proceeded with excellent enantioselectivity, although the expected trend of a decrease in conversion and enantioselectivity for the more bulky t-butyl substituent was also observed (Fig. 7). Also with the increase of the α-carbon chain (–CH2CH2CH3) (Fig. 7, VIII), the selectivity and activity of the reaction drops significantly. This result illustrates the importance of bulky groups next to the carbonyl for obtaining high enantioselectivity. The aromatic ketones (Fig. 7, VII) with a strong σ-electron withdrawing and weak π-electron donating chloro group and with the π-electron donating methoxy and methyl group in the para position showed a higher selectivity and lower activity compared to that with propiophenone. These results indicate that the para substituted ketones have major effect on the catalytic hydrogenation of ketones.
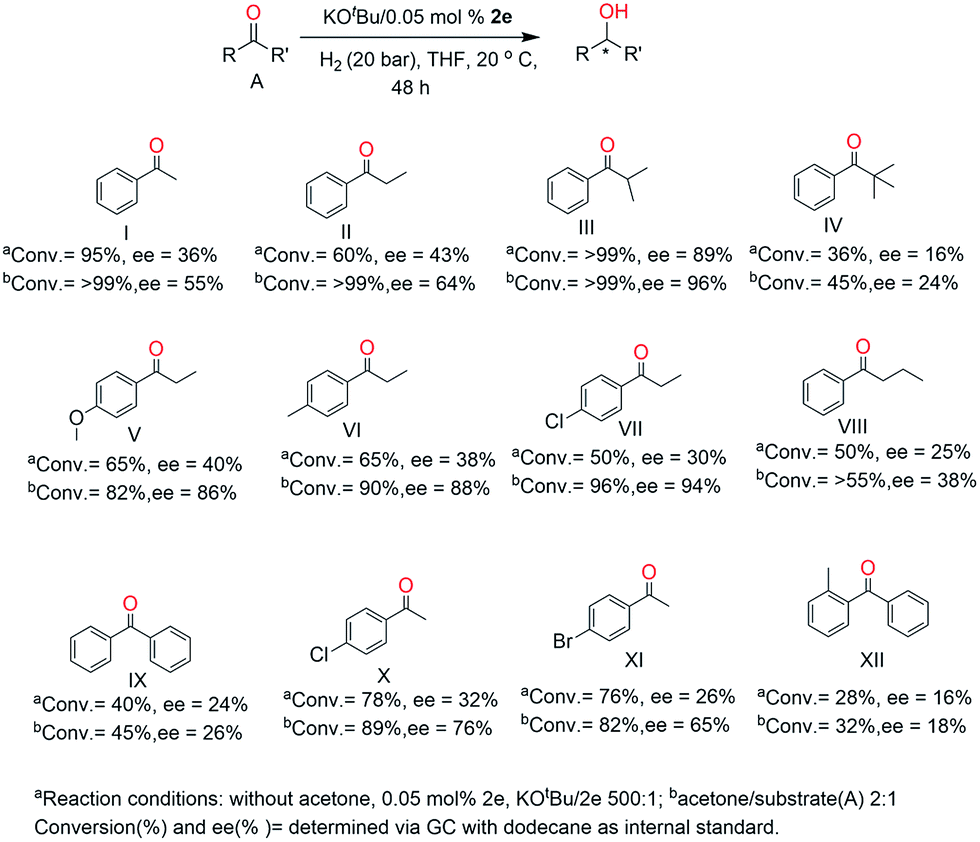 | ||
| Fig. 7 Asymmetric hydrogenation of ketones catalyzed by 2e. | ||
In contrast, the dinuclear iridium complexes 1a, b and 2c, d show lower catalytic activity and selectivity for the hydrogenation of simple ketones. For example asymmetric hydrogenation of α-methylpropiophenone catalyzed by 1a and 2c (0.05 mol%) in the presence of excess KOtBu and acetone under same reaction condition (see Table S2†) gave corresponding alcohol in 40% and 35% yield and 24% and 8% ee. While 1b and 2d gave 23% and 16% yield and 11% and 2.0% ee. Needless to say, no reaction occurred in the absence of KOtBu. This may suggest that dinuclear iridium complexes act as catalyst precursor to promote enantioselective asymmetric hydrogenation under the influence of base and acetone.
4. Conclusions
In conclusion the reported pyridylalkylamine mononuclear iridium complexes 2e–h represents a novel class of efficient and easily accessible catalysts for the asymmetric hydrogenation of simple ketones. Due to the modular ligand design, broad substitution patterns can be realized. The moderate to high efficiency and good selectivity combined with the novel structural motif opens up new prospects for the enantioselective hydrogenation of ketones.Acknowledgements
Financial support from Deutsche Forschungsgemeinschaft is gratefully acknowledged. P. Kumar is thankful to the Deutscher Akademischer Austausch Dienst (DAAD) for a fellowship.Notes and references
- (a) R. M. Bullock, Chem.–Eur. J., 2004, 10, 2366 CrossRef CAS PubMed; (b) R. M. Bullock, in Handbook of Homogeneous hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007, ch. 7 Search PubMed; (c) R. Noyori and T. Ohkuma, Angew. Chem., 2001, 113, 40 CrossRef; (d) R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40 Search PubMed; (e) N. B. Johnson, I. C. Lennon, P. H. Moran and J. A. Ramsden, Acc. Chem. Res., 2007, 40, 1291 CrossRef CAS PubMed.
- W. S. Knowles, Angew. Chem., Int. Ed., 2002, 41, 1998 CrossRef CAS.
- W. S. Knowles and R. Noyori, Acc. Chem. Res., 2007, 40, 1238 CrossRef CAS PubMed.
- R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008 CrossRef CAS PubMed.
- (a) N. B. Johnson, I. C. Lennon, P. H. Moran and J. A. Ramsden, Acc. Chem. Res., 2007, 40, 1291 CrossRef CAS PubMed; (b) C. S. Schultz and S. W. Krska, Acc. Chem. Res., 2007, 40, 1320 CrossRef PubMed; (c) H. Shimizu, I. Nagasaki, K. Matsumura, N. Sayo and T. Saito, Acc. Chem. Res., 2007, 40, 1385 CrossRef CAS PubMed.
- (a) R. Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley, New York, 1994, Search PubMed; (b) R. Noyori, Chem. Soc. Rev., 1989, 18, 187 RSC; (c) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS.
- (a) P. Braunstein, F. Naud and S. J. Rettig, New J. Chem., 2001, 25, 32 RSC; (b) P. Braunstein, M. D. Fryzuk, F. Naud and S. J. Rettig, J. Chem. Soc., Dalton Trans., 1999, 589 RSC; (c) P. Braunstein, F. Naud, C. Graiff and A. Tiripicchio, Chem. Commun., 2000, 897 RSC.
- W. A. Herrmann and B. Cornils, Angew. Chem., Int. Ed., 1997, 36, 1049 CAS.
- (a) R. H. Crabtree, H. Felkin and G. E. Morris, J. Organomet. Chem., 1977, 135, 205 CrossRef; (b) R. H. Crabtree and G. E. Morris, J. Organomet. Chem., 1977, 135, 395 CrossRef CAS; (c) R. H. Crabtree, Acc. Chem. Res., 1979, 12, 331 CrossRef CAS; (d) R. H. Crabtree, H. Felkin, T. Fillebeen-Khan and G. E. Morris, J. Organomet. Chem., 1979, 168, 183 CrossRef CAS.
- Examples of phosphorus-free ruthenium catalysts: (a) M. Ito, M. Hirakawa, K. Murata and T. Ikariya, Organometallics, 2001, 20, 379 CrossRef CAS; (b) I. Karame, M. Jahjah, A. Messaoudi, M. L. Tommasino and M. Lemaire, Tetrahedron: Asymmetry, 2004, 15, 1569 CrossRef CAS; (c) T. Ohkuma, N. Utsumi, K. Tsutsumi, K. Murata, C. A. Sandoval and R. Noyori, J. Am. Chem. Soc., 2006, 128, 8724 CrossRef CAS PubMed; (d) C. A. Sandoval, T. Ohkuma, N. Utsumi, K. Tsutsumi, K. Murata and R. Noyori, Chem.–sian J., 2006, 1–2, 102 Search PubMed; (e) M. L. Tommasino, C. Thomazeau, F. Touchard and M. Lemaire, Tetrahedron: Asymmetry, 1999, 10, 1813 CrossRef CAS; (f) M. L. Tommasino, M. Casalata, J. A. Breuzard, M. C. Bonnet and M. Lemaire, Stud. Surf. Sci. Catal., 2000, 130D, 3369 CrossRef CAS; (g) C. Maillet, T. Praveen, P. Janvier, S. Minguet, M. Evain, C. Saluzzo, M. L. Tommasino and B. Bujoli, J. Org. Chem., 2002, 67, 8191 CrossRef CAS PubMed; (h) J. Liu, X. Wu, X. Wu, J. A. Iggo and J. Xiao, Coord. Chem. Rev., 2008, 252, 782 CrossRef CAS; (i) J. E. D. Martins, D. J. Morris and M. Wills, Tetrahedron Lett., 2009, 50, 688 CrossRef CAS.
- (a) X. Wu, C. Corcoran, S. Yang and J. Xiao, ChemSusChem, 2008, 1, 7 CrossRef PubMed; (b) S.-I. Inuoe, K. Nomura, S. Hashiguchi, R. Noyori and Y. Izawa, Chem. Lett., 1987, 957 Search PubMed.
- (a) R. Noyori, Adv. Synth. Catal., 2003, 345, 15 CrossRef CAS; (b) R. Noyori, C. A. Sandoval, K. Muniz and T. Ohkuma, Philos. Trans. R. Soc. London, Ser. A, 2005, 363, 901 CrossRef CAS PubMed; (c) C. Hedberg, in Modern Reduction Methods, ed. P. G. Andersson and I. J. Munslow, Wiley-VCH, 2008, ch. 5, Search PubMed; (d) R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2006, 40, 40 CrossRef.
- (a) H. Doucet, T. Ohkuma, K. Murata, T. Yokozawa, E. Katayama, A. F. England, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed., 1998, 37, 1703 CrossRef CAS; (b) T. Ohkuma, H. Ooka, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 2675 CrossRef CAS; (c) C. A. Sandoval, T. Ohkuma, K. Muniz and R. Noyori, J. Am. Chem. Soc., 2003, 125, 13490 CrossRef CAS PubMed; (d) R. Abbel, K. Abdur-Rashid, M. Faatz, A. Hadzovic, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2005, 127, 1870 CrossRef CAS PubMed; (e) T. Leysson, D. Peters and J. N. Harvey, Organometallics, 2008, 27, 1514 CrossRef.
- (a) V. Rautenstrauch, X. Hoang-Cong, R. Churland, K. Abdur-Rashid and R. H. Morris, Chem.–Eur. J., 2003, 9, 4954 CrossRef CAS PubMed; (b) J.-X. Gao, H. Zhang, X.-D. Yi, P.-P. Xu, C.-L. Tang, H.-L. Wan, K.-R. Tsai and T. Ikariya, Chirality, 2000, 12, 383 CrossRef CAS PubMed.
- (a) T. Li, R. Churland, A. J. Lough, K. Abdur-Rashid and R. H. Morris, Organometallics, 2004, 23, 6239 CrossRef CAS; (b) R. J. Hamilton and S. H. Bergens, J. Am. Chem. Soc., 2006, 128, 13700 CrossRef CAS PubMed; (c) S. E. Clapham, R. Guo, M. Zimmer-De Iuliis, N. Rasool, A. Lough and R. H. Morris, Organometallics, 2006, 25, 5477 CrossRef CAS; (d) R. Puchta, L. Dahlenburg and T. Clark, Chem.–Eur. J., 2008, 14, 8898 CrossRef CAS PubMed.
- C. S. Chin and B. Lee, J. Chem. Soc., Dalton Trans., 1991, 1323 RSC.
- L. A. Oro and J. A. Cabeza, Inorg. Synth., 1985, 23, 128 Search PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115 CrossRef CAS.
- G. M. Sheldrick, SHELX-97, Programm for Cystal Structure Analysis (Release 97-2), Institut für Anorganische Chemie der Universität Göttingen, Germany, 1998 Search PubMed.
- L. J. Farrudia, J. Appl. Crystallogr., 1999, 32, 873 Search PubMed.
- (a) S. Striegler and M. Dittel, Inorg. Chem., 2005, 44, 2728 CrossRef CAS PubMed; (b) J. M. Botha, K. Umakoshi, Y. Sasaki and G. J. Lamprecht, Inorg. Chem., 1998, 37, 1609 CrossRef CAS; (c) J. Chen, X. Wang, Y. Zhu, J. Lin, X. Yang, Y. Li, Y. Lu and Z. Guo, Inorg. Chem., 2005, 37, 3422 CrossRef PubMed.
- V. B. Birman, E. W. Uffman, H. Jiang, X. Li and C. J. Kilbane, J. Am. Chem. Soc., 2004, 126, 12226 CrossRef CAS PubMed.
- R. Uson, L. A. Oro and J. A. Cabeza, Inorg. Synth., 1985, 23, 126 CrossRef CAS.
- T. Irrgang, D. Friedrich and R. Kempe, Angew. Chem., Int. Ed., 2011, 50, 2183 CrossRef CAS PubMed.
- (a) A. Berkessel, T. J. S. Schubert and T. N. Muller, J. Am. Chem. Soc., 2002, 124, 8693 CrossRef CAS PubMed; (b) C. Walling and L. Bollyky, J. Am. Chem. Soc., 1964, 86, 3750 CrossRef CAS; (c) C. Walling and L. Bollyky, J. Am. Chem. Soc., 1961, 83, 2968 CrossRef CAS.
- R. Hartmann and P. Chen, Angew. Chem., 2001, 113, 3693 ( Angew. Chem., Int. Ed. , 2001 , 40 , 3581 ) CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1437480–1437484 and 1473406. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra04524c |
| This journal is © The Royal Society of Chemistry 2016 |