
Effect of RGO/ZnxCd1−xS crystalline phase on solar photoactivation processes†
Omran Moradlou*a,
Neda Tedadia,
Alireza Banazadehb and
Naimeh Naseric
aDepartment of Chemistry, Alzahra University, P.O. Box: 1993893973, Tehran, Iran. E-mail: moradlou@alzahra.ac.ir
bDepartment of Chemistry and Biochemistry, Texas Tech University, Lubbock, TX, USA
cDepartment of Physics, Sharif University of Technology, P.O. Box 115559161, Tehran, Iran
First published on 4th May 2016
Abstract
A series of reduced graphene oxide/ZnxCd1−xS (RGO/ZnxCd1−xS) nanocomposites (0 < x < 1) with different ratios of Zn/Cd were synthesized via a facile hydrothermal route under optimized experimental conditions and were carefully characterized by various techniques. Because very little is known about the morphology, specific surface area, and crystal phase effects of RGO/ZnxCd1−xS crystals on their photoresponsivity, field-emission scanning electron microscopy (FE-SEM), BET surface area analysis and X-ray diffraction (XRD) data were studied to investigate their effects on photoactivity. Based on the results, a crystal phase transition from a cubic phase in RGO/Zn0.9Cd0.1S to a hexagonal wurtzite phase in RGO/Zn0.8Cd0.2S nanocomposites occurs and the crystalline phase is the main factor influencing the photoresponsivity of RGO/ZnxCd1−xS nanocomposites in photodegradation and photoelectrochemical (PEC) processes under visible light irradiation. According to the results, the RGO/Zn0.8Cd0.2S nanocomposite with the hexagonal crystal phase revealed better photocatalytic activity (k = 9.4 × 10−3 min−1) and PEC response (∼2.0 mA cm−2) in comparison with the cubic crystal phase.
1. Introduction
As an ideal green technology, semiconductor photocatalysts have attracted continuous interest for their potential applications in environmental cleaning and solar energy utilization.1,2 So far, numerous active photocatalysts such as TiO2 have been developed for the photodegradation of organic pollutants.3,4 Chalcogenides such as CdS5,6 and ZnS7 are also regarded as good candidates for photocatalytic applications. But, the band gap energy (Eg) of ZnS is 3.6 eV, which is too large to respond under visible light8,9 and CdS suffers a photocorrosion reaction, resulting in loss of material during irradiation.10,11 Therefore, numerous attempts have been made in the preparation of ZnxCd1−xS (0 < x < 1) composites to shift the absorption edge of ZnS and to overcome the photocorrosion reaction of CdS.12,13 These ZnCdS-based composites have proved to be efficient visible-light driven photocatalysts and photoelectrochemical (PEC) photoanodes and have been designed to achieve an enhanced photocatalytic activity as well as hydrogen generation.14,15 Based on recent reports, the Eg of a ZnxCd1−xS nanostructure can be flexibly tuned by changing the molar ratio of Zn/Cd to achieve the best band gap to have a photoresponse in the visible region.16,17 The photocorrosion resistance, proper band structure, high photocatalytic function and chemical stability of ZnxCd1−xS cause these nanocomposites to be attractive photocatalysts and PEC photoanodes.In literature, mainly two classes of factors are reviewed affecting the photocatalytic activity of photocatalysts; the electronic factors including band gap energy, photoexcitation, bulk diffusion and surface transfer of carriers;18 and the morphological factors including the surface area and crystal/particle size.19 The photocatalytic reactions occur at the interface between catalyst surfaces and the adsorbed organic pollutants. So, although the photocatalytic (PC) reaction as a heterogeneous reaction is related to these factors, for design and introducing new photocatalysts, the structure and the crystal phase are also very crucial, and the photocatalytic activity strongly depends on the crystal phase structure as well as exposed facets of a crystal.20,21 Interestingly, the crystalline phase determines the crystal structure, electronic structure and surface atomic geometry22 and thus will affect the photoinduced processes.
The morphology, crystalline phase and crystal-facet-controlled fabrication of semiconductor materials have recently attracted considerable attention due to the fact that the photoelectric and photocatalytic properties of semiconducting materials can be enhanced by tailoring the surface atomic structures.23
Many studies in the investigation of morphology and structure effects on the photocatalytic activity are limited to TiO2,24,25 TiO2–ZnO hybrid,26 Ag3PO4,23 CdS,27 BiVO4,28 and alkaline niobates.29,30 Bi et al. have studied the crystal phase effects of Ag3PO4 crystals on their photocatalytic performance and reported that although the specific surface area of the samples are the same, Ag3PO4 with rhombic dodecahedrons exhibits much higher activities than cubes for the degradation of organic contaminants.23 BiVO4 photocatalysts with various crystalline phases including tetragonal, monoclinic and monoclinic/tetragonal heterophase were also synthesized by Fan et al.28 and their PC activities have been examined on the photodegradation of methylene blue. The results indicated that the photocatalytic performance of BiVO4 largely depends on the crystalline phases and the behaviour of photoinduced charge carriers, and the photocatalyst with monoclinic scheelite structure has much higher PC activity.
Although the photocatalytic and photoelectrochemical activities of ZnxCd1−xS nanostructured materials have been investigated recently, far less information is available regarding the effects of morphology and structure of ZnxCd1−xS. A large number of reports on ZnxCd1−xS photocatalysts have revealed that high efficiency of Zn1−xCdxS is a result of their appropriate width of band gap energy. However, besides the band structure and band gap width, the structural properties were also studied13 and was reported that the heterojunction formed by the coexistence of zinc blende and wurtzite phases in ZnCdS can significantly improve the separation and migration of electron–hole pairs. It should be noted that the effects of crystalline phase of reduced graphene oxide/ZnxCd1−xS (RGO/ZnxCd1−xS) on their photocatalytic activities were seldom reported and have received only sporadic attention. Therefore, it is necessary to make a thorough inquiry into the effect of crystal phase besides other factors such as specific surface area, band position and band gap width on their photoresponsivity.
Due to the fact that the morphology of the composites is accompanied by the surface area and crystal growth, the effects of morphology, specific surface area and crystal phase should be discussed together in detail. So, the main aim of this work is the investigation of the effect of these factors on the photoresponsivity of RGO/ZnxCd1−xS nanocomposites. A facile hydrothermal method was used for the synthesis of a series of RGO/ZnxCd1−xS nanocomposites by changing the molar ratio of Zn/Cd. During the hydrothermal synthesis process, ZnxCd1−xS (0 < x < 1) nanoparticles were grown on RGO surface. Then, the synthesized nanocomposites were characterized by various techniques including diffuse reflectance spectroscopy (DRS) to obtain the band gap energy, Brunauer–Emmett–Teller (BET) surface area analysis to determine the specific surface area and porosity, X-ray photoelectron spectroscopy (XPS) to measure the chemical state of the elements in the material and X-ray diffraction (XRD) to investigate the crystal phases and crystalline sizes of RGO/ZnxCd1−xS nanocomposites. Finally, their visible light responsive PC and PEC activities and role of their particular crystalline phase on photoresponsivity were investigated.
2. Experimental and methods
2.1 Materials
Graphite powder (purity 99.99%), potassium permanganate (KMnO4), sulfuric acid (H2SO4, 98%), hydrogen peroxide, sodium nitrate, zinc nitrate (Zn(NO3)2·4H2O), cadmium nitrate (Cd(NO3)2·4H2O), thiourea (CS(NH2)2), magnesium sulphate (MgSO4), sodium sulphide (Na2S) and methylene blue (C16H18ClN3S) were purchased from Merck. All reagents were of analytical grade and used without further purification.2.2 Preparation of graphene oxide and RGO/ZnxCd1−xS nanocomposites
Graphene oxide (GRO) was prepared by a modified Hummers' method.31 In a typical procedure, 0.50 g NaNO3 and 0.5 g graphite were put into 20 mL concentrated H2SO4 while it was cooled to 0 °C in an ice bath. Then, 3.00 g KMnO4 was slowly added into the flask under stirring conditions and allowed to reach the room temperature (25 ± 2 °C). The suspension was stirred continuously in a water bath for 2 h at 35 °C. Then, the prepared suspension was diluted by 40 mL of deionized (DI) water and kept in ice bath. During the dilution, the temperature of the suspension was maintained at less than 60 °C. After 15 min, the reaction was terminated by slow addition of 100 mL DI water along with 3.0 mL H2O2 (30%) to reduce the unreacted MnO4−. The obtained suspension was filtered and washed with DI water and HCl (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10). Subsequently, the suspension was placed into the ultrasonic bath for 30 min in order to exfoliate the graphite oxide layers to get graphene oxide nanosheets. Finally, the graphene oxide aqueous suspension was centrifuged with 3000 rpm for 10 min to remove the unexfoliated graphitic plates and remaining graphite particles.
:10). Subsequently, the suspension was placed into the ultrasonic bath for 30 min in order to exfoliate the graphite oxide layers to get graphene oxide nanosheets. Finally, the graphene oxide aqueous suspension was centrifuged with 3000 rpm for 10 min to remove the unexfoliated graphitic plates and remaining graphite particles.
A facile hydrothermal method was used to prepare RGO/ZnxCd1−xS nanocomposites by using the precursors of Zn(NO3)2·4H2O, Cd(NO3)2·4H2O, thiourea and synthesized graphene oxide. Different molar ratios of Zn/Cd (x = 0, 0.2, 0.5, 0.8, 0.9, 1) were applied in the preparation of nanocomposites by using of various amounts of Zn2+ (0.20 M), Cd2+ (0.20 M) and CS(NH2)2 (1.0 M) precursors. Graphene oxide (0.04 g) was added to solution with continuous stirring and sonication. Subsequently, this mixture was transferred into a 100 mL Teflon-lined autoclave and maintained at 180 °C for 24 h. Finally, the obtained product was filtered and washed by DI water for several times and dried at room temperature.
2.3 Photocatalytic activity
The photocatalytic activity of the synthesized samples were investigated by photodegradation of methylene blue (MB) aqueous solution under visible light irradiation using a 400 mW cm−2 Xe lamp equipped with a 400 nm cut-off filter. In all experiments, 20 mg of RGO/ZnxCd1−xS nanocomposite was dispersed in 100 mL of MB aqueous solution with the initial concentration of 1.0 × 10−5 M. Before irradiation, the suspension was magnetically stirred in dark conditions for 30 min to reach the adsorption–desorption equilibrium between photocatalyst–dye. Then, the mixture was placed into a 100 mL quartz cell, 30 cm away from the light source. During irradiation, 2.0 mL of the solution was sampled at regular intervals (20 min) for the analysis of MB concentration. The absorbance variations of MB were measured using a UV-vis spectrophotometer (Perkin Elmer, Lambda35) in the wavelength range of 200–800 nm.2.4 Photoelectrochemical measurements
At first, to fabricate a photoanode as the working electrode, the electrophoretic method was applied to deposit synthesized graphene oxide (GRO) nanosheets on a titanium (Ti) sheet (1.0 × 3.0 cm2) as a suitable substrate. During the electrophoretic deposition in solution containing GRO (1.0 mg mL−1) and MgSO4 (10 mg L−1), GRO is deposited on Ti sheet and reduced to RGO.32 Then, by using a feasible co-precipitation hydrothermal method which Ti sheet was immersed vertically in hydrothermal solution, Ti/RGO/ZnxCd1−xS photoanodes were fabricated. Photocurrent responses of photoanodes were investigated in a three-electrode electrochemical cell with photoanode as a working electrode, a platinum plate as a counter electrode, and an Ag/AgCl (KCl 3.0 M) as a reference electrode. The photoelectrochemical (PEC) measurements were carried out using a galvanostat/potentiostat Autolab PGSTAT101 instrument and a 400 mW cm−2 Xe lamp with UV cut-off filter (λoff < 400 nm) as the source of visible light irradiation. During all PEC measurements, 0.1 M Na2S was used as electrolyte.2.5 Instruments and material characterization
The morphology of the prepared photocatalysts was observed with field emission-scanning electron microscopy (FE-SEM, EM3900M, ZEISS, Germany) coupled with energy dispersive X-ray spectroscopy (EDS) for inspecting the elemental composition. The samples were coated with a thin layer of gold, prior to setting in the apparatus. The surface chemical composition of the synthesized samples was characterized by X-ray photoelectron spectroscopy (XPS). All binding energies were calibrated by taking the C 1s peak at 285.0 eV as a reference. The Fourier transform infrared (FT-IR) spectra were recorded on Bruker FT-IR Spectrometric Analyzer. The pellets of powder samples were made using potassium bromide (KBr) with the ratio of 1:50 (sample:KBr). To study the crystalline phase of synthesized RGO/ZnxCd1−xS nanocomposites, X-ray powder diffraction (XRD, PW1800, Philips) with Co Kα radiation (λ = 1.78 Å) was used. To determine the crystalline phase and size of the samples, X'Pert High Score software (version 1.0d) was applied. Brunauer–Emmett–Teller (BET) specific surface area (SBET) of the nanocomposites were investigated by nitrogen adsorption–desorption measurements at 77 K. In order to study the absorption spectra of samples (to calculate the Eg), UV-visible diffuse reflectance spectroscopy (UV-vis DRS, Avantes, Avaspec 2048) was used. BaSO4 was applied as a reflectance standard in DRS experiments.
3. Results and discussion
3.1 Physical characterizations
To achieve the aim of this work, morphology, surface area and crystalline phase of the prepared RGO/ZnxCd1−xS samples as well as their absorption edge and the energy band gap were analysed in detail to investigate the effects of these parameters on the PC and PEC photoresponsivity of the samples. | ||
| Fig. 1 FE-SEM images of RGO/ZnxCd1−xS nanocomposites, (a) RGO/Zn0.2Cd0.8S (b) RGO/Zn0.5Cd0.5S, (c) RGO/Zn0.8Cd0.2S, (d) RGO/Zn0.9Cd0.1S. | ||
The BET (Brunauer–Emmett–Teller) surface areas (SBET) and porous structures of the synthesized samples were investigated by nitrogen adsorption–desorption measurements. Adsorption equilibriums were displayed by isotherm curves regularly. Fig. 2a indicates the nitrogen adsorption–desorption isotherms of RGO/ZnxCd1−xS nanocomposites. These isotherms for all the samples are of type IV with H3 hysteresis loops in accordance with IUPAC classification indicating the formation of mesopores (2–50 nm) and the presence of uniform slit-like pores in samples.35,36 Isotherms of these nanocomposites show high adsorption at a high relative pressure (P/P0) ranging from 0.8 to 1.0. The Barrett–Joyner–Halenda (BJH) pore-size distribution curves (Fig. 2b) further confirm that all the prepared materials are mainly consist of mesopores in the wide range from 2 to 50 nm. After the analysis of the experimental data, the results showed that the specific surface area of RGO/ZnxCd1−xS nanocomposites increases with increase in Zn content probably due to decrease in sample density as reported elsewhere.16 According to the obtained results, RGO/Zn0.9Cd0.1S nanocomposite exhibits the largest specific surface area (40.70 m2 g−1) compared with other nanocomposites (Table 1).
 | ||
| Fig. 2 (a) Nitrogen adsorption–desorption isotherms for RGO/Zn0.9Cd0.1S, RGO/Zn0.8Cd0.2S and RGO/Zn0.5Cd0.5S. (b) BJH pore-size distribution curves of RGO/Zn0.9Cd0.1S, RGO/Zn0.8Cd0.2S and RGO/Zn0.5Cd0.5S. | ||
| Photocatalyst | Crystalline phase | Crystallite size (nm) | Specific surface area (m2 g−1) | Pore size (nm) |
|---|---|---|---|---|
| RGO/Zn0.9Cd0.1S | Cubic | 20.4 | 40.70 ± 1.50 | 9.183 |
| RGO/Zn0.8Cd0.2S | Hexagonal | 24.3 | 13.50 ± 1.10 | 12.542 |
| RGO/Zn0.5Cd0.5S | Hexagonal | 40.5 | 8.02 ± 0.70 | 19.188 |
| RGO/Zn0.2Cd0.8S | Hexagonal | 45.1 | 7.62 ± 0.80 | 21.052 |
To obtain the elemental composition of synthesized nanocomposites, energy dispersive X-ray spectroscopy (EDS) measurements were performed demonstrating the existence of Zn, Cd, C, and S elements in the as-prepared nanocomposites (Fig. S1, ESI†).
 | ||
| Fig. 3 (a) XRD patterns of the RGO/ZnxCd1−xS nanocomposites, (b) Raman spectra of GRO and RGO. | ||
For RGO/Zn0.9Cd0.1S sample, however, XRD pattern shows three main diffraction peaks at 2θ of 33.3068°, 55.7712° and 66.199° assigning to (111), (220) and (311) crystal planes of cubic sphalerite Zn0.9Cd0.1S nanostructure (JCPDS 24-1137), respectively. Interestingly, these results show that the crystalline phase of the samples transfers from hexagonal wurtzite to cubic phase in RGO/ZnxCd1−xS when the Zn content increases from x = 0.8 to 0.9. This is in good agreement with the results reported earlier16 and the results reveal that by changing the Zn/Cd ratio, the structure phase transition occurs and the crystal phase of RGO/ZnxCd1−xS samples can be controlled through adjusting the Zn/Cd ratio. For comparison, the XRD pattern of the pure Zn0.8Cd0.2S sample without graphene is also shown in Fig. 3a. The pattern is similar to that of RGO/Zn0.8Cd0.2S and the diffraction peak related to RGO (2θ = 25.56°)37 disappeared.
This is probably due to the low diffraction intensity of RGO, its low amount in samples or even its overlapping with a main peak of Zn0.8Cd0.2S hexagonal phase.3,38
As it is clear, from the XRD patterns, the diffraction peaks of samples gradually shift to larger angles with increase in Zn content. The angle related to main peak increases from 30.7911 in RGO/Zn0.5Cd0.5S to 33.3068 in RGO/Zn0.9Cd0.1S nanocomposite, indicating the incorporation of Zn2+ into the CdS lattice.39 The crystalline lattice distance (d-spacing) of CdS is decreased due to smaller radius of Zn2+ ion (0.74 Å) in comparison with Cd2+ (0.97 Å).16,40 The d-spacing values for RGO/Zn0.2Cd0.8S, RGO/Zn0.5Cd0.5S, RGO/Zn0.8Cd0.2S and RGO/Zn0.9Cd0.1S were obtained to be 3.3845, 3.3712, 3.3472 and 3.1235 Å, respectively. The peak shifts indicate that the synthesized samples are not a simple mixture of ZnS–CdS compounds and the nanocomposites of RGO/ZnxCd1−xS are formed.41,42 The average crystal sizes of RGO/ZnxCd1−xS samples were calculated by Scherrer equation and obtained to be 45.1, 40.5, 24.3 and 20.4 nm for x = 0.2, 0.5, 0.8 and 0.9, respectively (Table 1). Data shows that upon increasing Zn2+ content in RGO/ZnxCd1−xS samples, the mean grain size of composite decreases, mainly due to the smaller radius of Zn2+ in comparison with Cd2+.
Raman spectroscopy is widely used to determine the structure, defects and disorders of graphene. Fig. 3b indicates the Raman spectra of graphene oxide (GRO) and reduced graphene oxide (RGO). RGO shows three main peaks. The most intensity peak located at 1313 cm−1 correspond to D band of graphene which shows the presence of structure defects in graphene and is breathing mode of A1g symmetry containing K-point phonon.43 The other peak around 1594 cm−1 is related to G band of graphene that originates first-order scattering of E2g phonon for sp2 carbon. A small peak at around 2700 cm−1 represent 2D band of graphene that is due to two phonon double resonance Raman process. Raman spectrum of graphene oxide in Fig. 3b show the D and G band of GRO at about 1311 cm−1 and 1590 cm−1, respectively. As a result of the reduction of GRO to RGO, D and G band of GRO shift to lower values.44 The intensity of the D band in RGO is higher than that of GRO indicating the formation of more in-plane sp2 domains. The intensity ratio (ID/IG) of RGO is about 1.21 that is larger than that of GRO (1.14) indicating a reduction in the average size of the sp2 domains due to reduction of graphene oxide.44
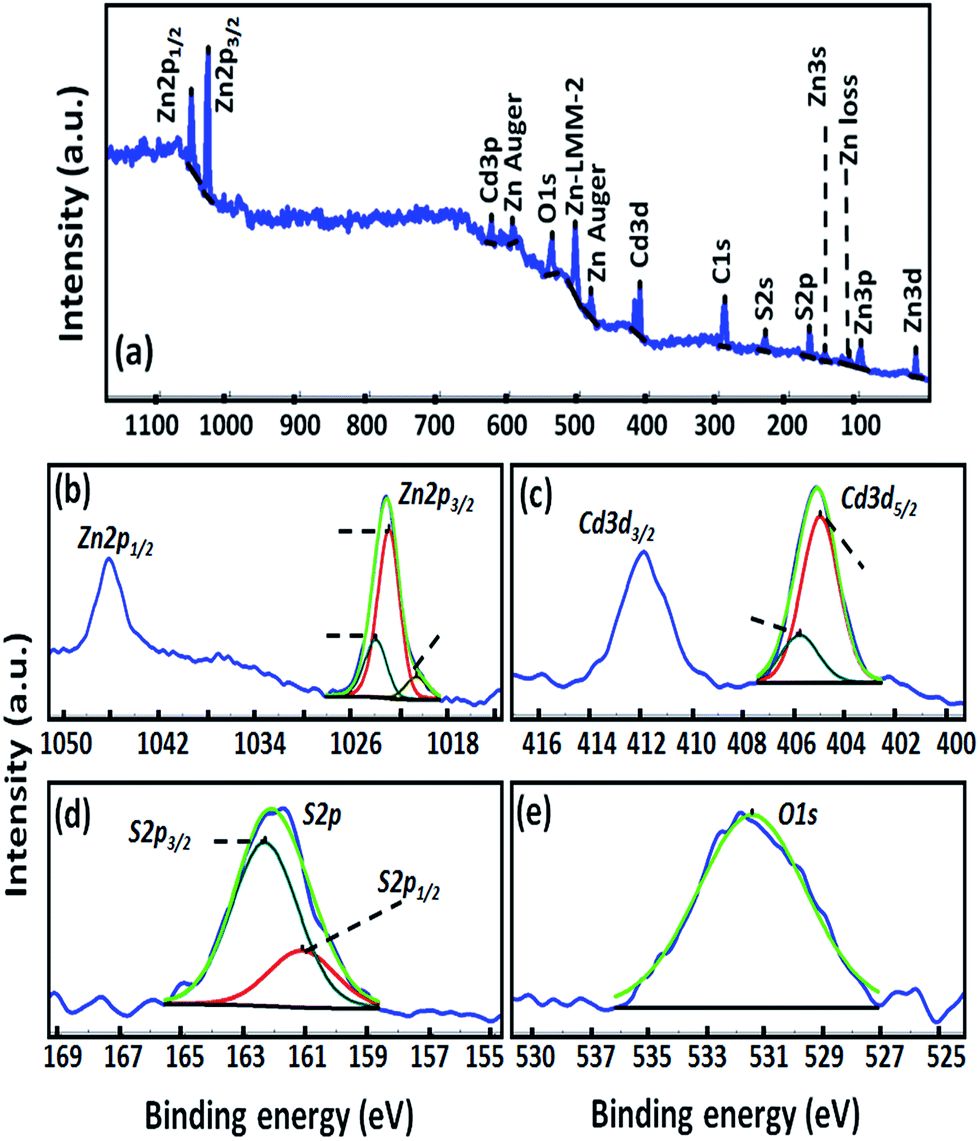 | ||
| Fig. 4 XPS spectra for RGO/Zn0.8Cd0.2S, (a) survey spectrum, (b) high resolution XPS window for Zn 2p, (c) Cd 3d, (d) S 2p, (e) O 1s. | ||
As shown, no impurities were found on the sample surface indicating the formation of pure and clean nanocomposite. The presence of C and O in the survey spectra is due to graphene and the adsorbed water molecules on the surface of nanocomposite, respectively. Fig. 4b displays the narrow scan XPS in Zn 2p range for RGO/Zn0.8Cd0.2S sample. Because of the spin–orbit coupling, Zn 2p is divided to Zn 2p3/2 and Zn 2p1/2 that are located at bonding energies of 1023 eV and 1046 eV, respectively. Data fitting was done on Zn 2p3/2 peak due to the higher intensity of this peak as compared with Zn 2p1/2.40 Zn 2p3/2 peak contains three separate peaks i.e. A, B and C that are related to Zn–S bond (1022.80 eV), Zn–OH bond (1023.90 eV) and metallic Zn (1020.50 eV), respectively. As shown in Fig. 4b, the major peak is for Zn–S bond (67.6%) which confirms the formation of Zn–S. The XPS spectra of Cd 3d was plotted in Fig. 4c including two different peaks. The binding energy of Cd 3d5/2 and Cd 3d3/2 for Zn0.8Cd0.2S sample appeared at 405.40 eV and 412.00 eV, respectively.40 Cd 3d5/2, as the most intense peak was separated into two peaks, A and B, which can be ascribed to the Cd–S bond (405.00 eV) and Cd–OH bond (405.80 eV). Similar to Zn 2p, Cd 3d5/2 major peak is located at bonding energy of 405.00 eV, which is assigned to Cd–S bond (77.7%) revealing the formation of CdS in the nanostructure. The positions of S 2p1/2 and S 2p3/2 high resolution XPS signals for RGO/Zn0.8Cd0.2S sample that are shown in Fig. 4d (A and B) are at the binding energies of 161.10 and 162.30 eV, respectively.45 It indicates that sulphur exists in the form of S2−. As mentioned above, Zn–OH and Cd–OH was slightly observed on the sample surface in XPS spectra related to Zn 2p and Cd 3d. These OH functional groups depict well itself under the oxygen peak in Fig. 4e. The position of oxygen peak was observed at 531.47 eV, which corresponds to OH groups accumulated on the surface of sample which is attributed to the formation of Zn and Cd hydroxides as reported before.46 By measuring the surface surrounded under XPS peak of each element, surface concentration of Zn, S, O and Cd have been estimated and listed in Table 2.
| Composition | O | Zn | Cd | S | Zn(ZnS) | Zn(Zn(OH)2) | Cd(CdS) | Cd(Cd(OH)2) |
| Surface concentration (%) | 30.7 | 46.8 | 4.1 | 18.4 | 67.6 | 23.1 | 77.7 | 22.3 |
To calculate the electronic states and the band gap energy (Eg) of the samples, diffuse reflectance spectra (DRS) of the samples were investigated. Eg of RGO/ZnxCd1−xS samples were calculated by a plot of (αhν)2 vs. photon energy (hν) based on the Kubelka–Munk function (αhν = A(hν − Eg)n).47 The calculated Eg are depicted in Table 3. As it is clear, Eg of RGO/ZnxCd1−xS samples gradually increases from 2.50 eV to 3.11 eV by the increase in Zn content. So, the band gap energy of RGO/ZnxCd1−xS nanocomposites can be controlled by changing the molar ratio of Zn/Cd in precursors during the hydrothermal synthesis.
| Photocatalyst | Absorption edge wavelength (nm) | Band gap energy (eV) |
|---|---|---|
| RGO/ZnS | 340 | 3.66 |
| RGO/Zn0.9Cd0.1S | 400 | 3.11 |
| RGO/Zn0.8Cd0.2S | 440 | 2.84 |
| RGO/Zn0.5Cd0.5S | 470 | 2.65 |
| RGO/Zn0.2Cd0.8S | 490 | 2.50 |
| RGO/CdS | 530 | 2.34 |
Fourier transform infrared (FT-IR) spectra of graphene oxide and RGO/Zn0.8Cd0.2S was also studied (Fig. S2†). The broad peak at 3421 cm−1 is attributed to the stretching and bending vibration of OH groups of graphene oxide and the peaks at 1633 cm−1 and 1718 cm−1 can be assigned to the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO, respectively. The peaks at 1401 cm−1 and 1460 cm−1 correspond to the stretching vibrations of C–OH of carboxyl groups48 and the sharp peak at 1095 cm−1 is related to the C–O stretching vibrations.38 Comparing the peak intensities in FT-IR spectra of RGO/Zn0.8Cd0.2S and GRO confirms the reduction of graphene oxide to graphene during the hydrothermal synthesis.
C and CO, respectively. The peaks at 1401 cm−1 and 1460 cm−1 correspond to the stretching vibrations of C–OH of carboxyl groups48 and the sharp peak at 1095 cm−1 is related to the C–O stretching vibrations.38 Comparing the peak intensities in FT-IR spectra of RGO/Zn0.8Cd0.2S and GRO confirms the reduction of graphene oxide to graphene during the hydrothermal synthesis.
3.2 Photocatalytic activity of the RGO/ZnxCd1−xS nanocomposites
To investigate the effects of various parameters including morphology, specific surface area, pore size diameter, energy band gap (Eg) and the crystal phase of RGO/ZnxCd1−xS nanocomposites on their photoactivity, the photodegradation of methylene blue (MB) as a model compound under the visible light irradiation was studied. The kinetics of the degradation reaction can be described by pseudo first-order equation, ln(C0/C) = kt, where C0 and C are the initial concentration and concentration at time t of MB, respectively, and k denotes the photodegradation reaction rate constant. The C0/C vs. t and ln(C0/C) vs. t plots were presented in Fig. 5. Among RGO/ZnxCd1−xS samples, the photoactivity of RGO/CdS and RGO/ZnS samples were also investigated. RGO/CdS sample exhibited the highest photodegradation reaction rate constant (k = 1.09 × 10−2 min−1), mainly due to its suitable band gap energy (Eg of 2.4 eV) and high charge carrier (e− and h+) mobility (340 and 50 cm2 V−1 s−1 for e− and h+, respectively49,50) compared to a lot of photoactive semiconducting materials. But, because of the photocorrosion51 and fast recombination of generated charge carriers,52 CdS is not considered as a suitable photocatalyst. For RGO/ZnS, however, the k value was low (k = 6.6 × 10−3 min−1) due to its broad Eg and low photoactivity in visible region. The obtained photodegradation rate constants of RGO/ZnxCd1−xS nanocomposites were 9.4 × 10−3, 8.4 × 10−3, 7.9 × 10−3 and 7.5 × 10−3 min−1, for RGO/Zn0.8Cd0.2S, RGO/Zn0.5Cd0.5S, RGO/Zn0.2Cd0.8S and RGO/Zn0.9Cd0.1S, respectively. From these results, it is clear that the photocatalytic activity of RGO/ZnxCd1−xS is altered by varying the Cd/Zn ratio as reported previously.53 As Fig. 5 shows, when Cd content decreases, the photodegradation efficiency of MB for RGO/ZnxCd1−xS becomes higher (x = 0.2, 0.5) and reaches the highest for x = 0.8. These results could be attributed to the variation in conduction band (CB) edge position to the moderate and proper position,42 increase in specific surface area and pore size (Table 1). However, further decrease in Cd content, leads to a fast deterioration of PC efficiency for x = 0.9. In fact, the highest surface area in RGO/Zn0.9Cd0.1S did not endow it the best photoactivity. It means that the crystalline phase is the most important factor responsible for PC activity of RGO/ZnxCd1−xS nanocomposites in comparison with band gap, CB edge position, specific surface area and pore size. Variation in k values (min−1) is as follows: RGO/Zn0.8Cd0.2S (9.4 × 10−3) > RGO/Zn0.5Cd0.5S (8.4 × 10−3) > RGO/Zn0.2Cd0.8S (7.9 × 10−3) > RGO/Zn0.9Cd0.1S (7.5 × 10−3). From these data, the efficient photoactive sample is RGO/Zn0.8Cd0.2S.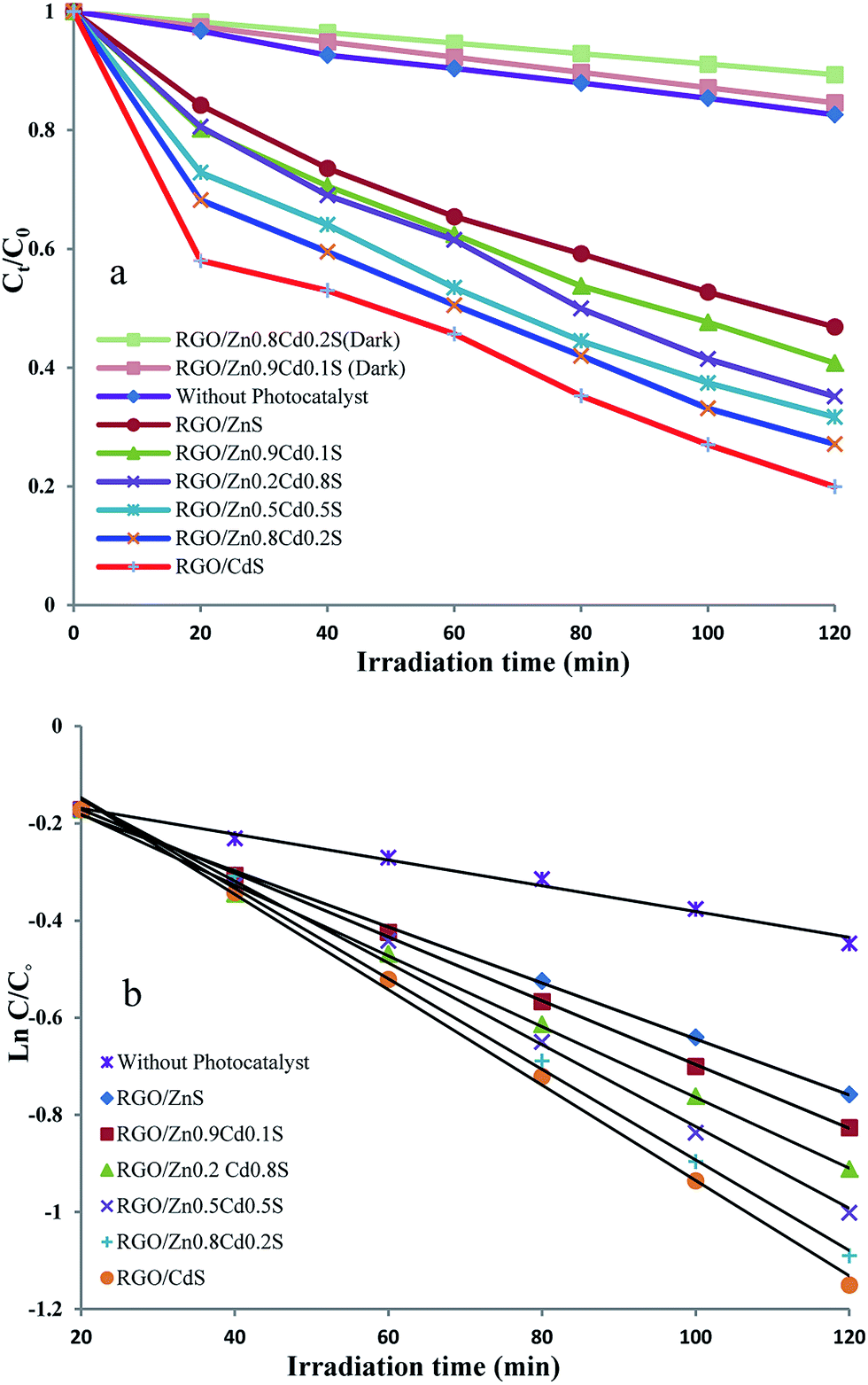 | ||
| Fig. 5 (a) Ct/C0 versus time curves of photodegradation of MB by RGO/ZnxCd1−xS photocatalysts. (b) The kinetics of photodegradation of MB by RGO/ZnxCd1−xS photocatalysts. | ||
According to the data presented in Table 1 and from comparing these data with the obtained k values, it would be concluded that the surface characteristics of the samples such as specific surface area and pore size are not the main parameters affecting the photoactivity of RGO/ZnxCd1−xS samples. The specific surface area of RGO/Zn0.8Cd0.2S with higher photocatalytic activity is about 3 times lower than that of RGO/Zn0.9Cd0.1S. Concerning only the specific surface area, RGO/Zn0.9Cd0.1S (with higher SBET) should exhibit higher photodegradation reaction rate than other synthesized RGO/ZnxCd1−xS samples. Nanocomposites with higher surface area are beneficial for higher charge carrier generation as they have more active sites upon light harvesting, and as a result, more reactions sites. But we have observed an opposite situation. Although the specific surface area of the RGO/Zn0.9Cd0.1S is the highest, its photoactivity is the lowest. Therefore, specific surface area is not the only decisive factor influencing the photo-activity of the samples. The pore size and crystal size of the samples show a similar trend from x = 0.2 to x = 0.9 (Table 1) and decrease by increase in x. The effect of particle size and specific surface area of the photocatalyst on the photodegradation rate (k) has been extensively studied (see for example ref. 54) and shown that the increase in surface area increases k. Finally, although the band gap energy of RGO/ZnxCd1−xS increases by increase in x from 0 to 1 (Table 3), the difference between Eg or absorption edge of RGO/Zn0.8Cd0.2S and RGO/Zn0.9Cd0.1S is not so high to affect the photoresponsivity of the sample considerably. So, from these findings, it seems that these parameters are not the main factors affecting the photoactivity of RGO/ZnxCd1−xS samples. Among these various factors, the crystalline phase is seems to be a major factor in the improvement of photocatalytic performance of RGO/ZnxCd1−xS nanocomposites. RGO/ZnxCd1−xS nanocomposite with hexagonal crystal phase revealed better photocatalytic activity in comparison with cubic crystal phase.
As reported previously,42 RGO increases the photoactivity of ZnxCd1−xS photocatalysts. In fact, RGO in RGO/ZnxCd1−xS composites act as an electron collector and transporter to effectively lengthen the lifetime of the charge carriers and to efficiently separate the photogenerated electron–hole pairs.55 So, graphene improves the charge separation in RGO/ZnxCd1−xS. As discussed in growth mechanism (Section 3.1.1), Zn2+ and Cd2+ cations tend to adsorb on the surface of GRO through electrostatic attractions and ZnxCd1−xS clusters would be nucleate and grow on graphene, and graphene would be as a conductive substrate for ZnxCd1−xS nanoparticles. So, there is an intimate interfacial contact between RGO and ZnxCd1−xS nanoparticles and such a good interfacial contact for RGO–ZnxCd1−xS, favours the charge carrier transfer process. So, under the illumination, the photogenerated electrons in CB of ZnxCd1−xS can be easily transferred to RGO, leading to the electron–hole separation. Consequently, graphene improves the charge separation and PC activity.
3.3 Photoelectrochemical activity of the RGO/ZnxCd1−xS
To investigate the photosensitivity of the prepared samples, the photocurrent density was measured in both dark and visible light illuminating conditions as a function of the applied potential of 0.5 V as bias (Fig. 6a).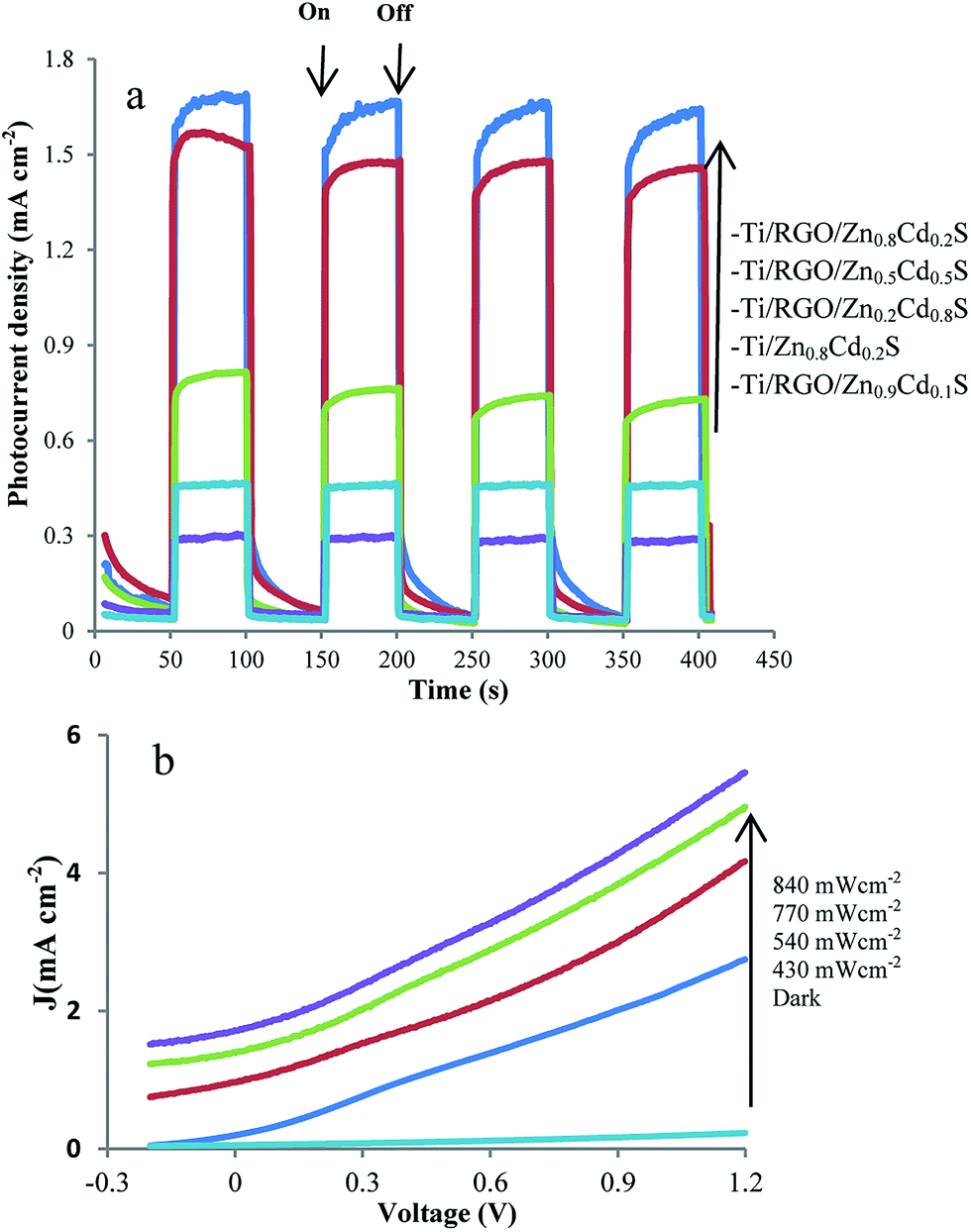 | ||
| Fig. 6 (a) The transient photocurrent density responses of Ti/RGO/ZnxCd1−xS and Ti/Zn0.8Cd0.2S in Na2S solution at a voltage bias of 0.5 V. (b) LSV of Ti/RGO/Zn0.8Cd0.2S photoanode in dark and different illumination intensities. | ||
The photoelectrochemical (PEC) performance of the samples was investigated by fabricating their corresponding photoanode with a titanium sheet as a substrate coated with ZnxCd1−xS and RGO/ZnxCd1−xS via hydrothermal method.
Our findings on PEC behaviour of various Ti/ZnxCd1−xS and Ti/RGO/ZnxCd1−xS photoanodes were in good agreement with the previous studies,14 indicating that the RGO/Zn0.8Cd0.2S nanocomposites has higher photoresponsivity in comparison with other RGO/ZnxCd1−xS nanocomposites. From the results (Fig. 6a), PEC photocurrent density of RGO/ZnxCd1−xS is altered by varying the Cd/Zn ratio. As Cd content decreases, the photocurrent density of RGO/ZnxCd1−xS becomes higher (x = 0.2, 0.5) and reaches the highest for x = 0.8. However, further decrease in Cd content, leads to a fast deterioration of PEC photocurrent for x = 0.9. As discussed for PC activity, these results indicated that the crystalline phase is the most important factor responsible for PC activity of RGO/ZnxCd1−xS nanocomposites in comparison with band gap, CB edge position and specific surface area.
Linear sweep voltammetry (LSV) was also used to investigate the photoresponsivity of Ti/RGO/Zn0.8Cd0.2S photoanode as a function of applied voltages from −0.2 to 1.2 V in dark and different illumination intensities (Fig. 6b). Based on the results of the photoelectrochemical activities, PEC of RGO/Zn0.2Cd0.8S with the hexagonal crystalline phase has the highest photoactivity and it can be concluded that the crystalline phase is again the major factor in the improvement of PEC performance. Previously reported works have indicated that the crystal phase influences the PEC behaviour of other photoactive semiconductors (see for example ref. 30 and 56). The effect of graphene in the improvement of photocurrent densities in fabricated photoanodes was evident. For comparison, the photoresponse of Ti/Zn0.8Cd0.2S at a bias potential of 0.5 was also shown in Fig. 6a. For both electrodes, upon the illumination of the photoanode surface, rapid generation of charge carriers is occurred. The generated photocurrent density of Ti/RGO/Zn0.8Cd0.2S is much higher than that of Ti/Zn0.8Cd0.2S mainly due to the effective enhancement in the photogenerated electron lifetime, efficient separation of the photogenerated electron–hole pairs and suppression in charge recombination rate in graphene based nanocomposites.55,57
4. Conclusions
RGO/ZnxCd1−xS nanocomposites with different molar ratios of Zn/Cd (x = 0, 0.2, 0.5, 0.8, 0.9, 1) were synthesized via a facile hydrothermal method in 180 °C for 24 h and were characterized by various techniques including FE-SEM, EDS, BET, XPS, DRS, FT-IR and XRD. BET analysis confirmed the higher specific surface area for RGO/Zn0.9Cd0.1S while its PC and PEC photosensitivity was lower than that of other synthesized RGO/ZnxCd1−xS samples. According to the XRD analysis, crystalline phase transition occurs from cubic phase in RGO/Zn0.9Cd0.1S to hexagonal wurtzite phase in RGO/Zn0.8Cd0.2S, and their photoactivity studies (including photocatalytic and photoelectrochemical) revealed that the photoresponsivity of the nanocomposites with hexagonal phase is higher than that of cubic phase.Acknowledgements
The authors would like to thank Iran National Science Foundation (Grant no. 91042597) for supporting the project.Notes and references
- Y. Wang, Q. Wang, X. Zhan, F. Wang, M. Safdar and J. He, Nanoscale, 2013, 5, 8326–8339 RSC.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- A. Kubacka, M. Fernandez-Garcia and G. Colón, Chem. Rev., 2012, 3, 1555–1614 CrossRef PubMed.
- T. Simon, N. Bouchonville, M. J. Berr, A. Vaneski, A. Adrovic, D. Volbers, R. Wyrwich, M. Doblinger, A. S. Susha, A. L. Rogach, F. Jackel, J. K. Stolarczyk and J. Feldmann, Nat. Mater., 2014, 13, 1013–1019 CrossRef CAS PubMed.
- M. Luo, Y. Liu, J. Hu, H. Liu and J. Li, ACS Appl. Mater. Interfaces, 2012, 4, 1813–1821 CAS.
- Q. Mi, J. Hu, M. Luo, Z. Huang and J. Li, Sci. Adv. Mater., 2013, 5, 1649–1657 CrossRef CAS.
- J. Zhang, J. G. Yu, Y. M. Zhang, Q. Li and J. R. Gong, Nano Lett., 2011, 11, 4774–4779 CrossRef CAS PubMed.
- B. N. Patil and S. A. Acharya, Adv. Mater. Lett., 2014, 5, 113–116 CAS.
- N. Soltani, E. Saion, M. Z. Hussein, M. Erfani, A. Abedini, G. Bahmanrokh, M. Navasery and P. Vaziri, Int. J. Mol. Sci., 2012, 13, 12242–12258 CrossRef CAS PubMed.
- Z. Khan, T. R. Chetia, A. K. Vardhaman, D. Barpuzary, C. V. Sastri and M. Qureshi, RSC Adv., 2012, 2, 12122–12128 RSC.
- X. Zong, H. J. Yan, G. P. Wu, G. J. Ma, F. Y. Wen, L. Wang and C. Li, J. Am. Chem. Soc., 2008, 130, 7176–7177 CrossRef CAS PubMed.
- X. Wang, B. Yuan, Z. Xie, D. Wang and R. Zhang, J. Colloid Interface Sci., 2015, 446, 150–154 CrossRef CAS PubMed.
- Y. Y. Hsu, N. T. Suen, C. C. Chang, S. F. Hung, C. L. Chen, T. S. Chan, C. L. Dong, C. C. Chan, S. Y. Chen and H. M. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 22558–22569 CAS.
- J. Zhang, J. Yu, M. Jaroniec and J. R. Gong, Nano Lett., 2012, 12, 4584–4589 CrossRef CAS PubMed.
- H. Liu, M. Luo, J. Hu, X. Zhou and J. Li, Sci. Adv. Mater., 2013, 5, 1157–1167 CrossRef CAS.
- Q. Li, H. Meng, P. Zhou, Y. Zheng, J. Wang, J. Yu and J. R. Gong, ACS Catal., 2013, 3, 882–889 CrossRef CAS.
- J. G. Yu, J. Zhang and M. Jaroniec, Green Chem., 2010, 12, 1611–1614 RSC.
- G. Liu, X. Wang, Z. Chen, H. M. Cheng and G. Q. Lu, J. Colloid Interface Sci., 2009, 329, 331–338 CrossRef CAS PubMed.
- A. L. Linsebigler, G. Q. Lu and J. J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- H. G. Yang, C. H. Sun, S. Z. Zou, J. Qiao, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–642 CrossRef CAS PubMed.
- E. S. Jang, J. H. Won, S. J. Hwang and J. H. Choy, Adv. Mater., 2006, 18, 3309–3312 CrossRef CAS.
- T. Vossmeyer, L. Katsikas, M. Giersig, I. G. Popovic, K. Diesner, A. Chemseddine, A. Eychmueller and H. Weller, J. Phys. Chem., 1994, 98, 7665–7673 CrossRef CAS.
- Y. Bi, S. Ouyang, N. Umezawa, J. Cao and J. Ye, J. Am. Chem. Soc., 2011, 133, 6490–6492 CrossRef CAS PubMed.
- J. Zhang, Q. Xu, Z. Feng, M. Li and C. Li, Angew. Chem., Int. Ed., 2008, 47, 1766–1769 CrossRef CAS PubMed.
- H. Cheng, J. Wang, Y. Zhao and X. Han, RSC Adv., 2014, 4, 47031–47038 RSC.
- C. Cheng, A. Amini, C. Zhu, Z. Xu, H. Song and N. Wang, Sci. Rep., 2014, 4, 4181 Search PubMed.
- D. Lang, Q. Xiang, G. Qiu, X. Feng and F. Liu, Dalton Trans., 2014, 43, 7245–7253 RSC.
- H. Fan, T. Jiang, H. Li, D. Wang, L. Wang, J. Zhai, D. He, P. Wang and T. Xie, J. Phys. Chem. C, 2012, 116, 2425–2430 CAS.
- L. Yan, J. Zhang, X. Zhou, X. Wu, J. Lan, Y. Wang, G. Liu, J. Yu and L. Zhi, Int. J. Hydrogen Energy, 2013, 38, 3554–3561 CrossRef CAS.
- P. Li, S. Ouyang, G. Xi, T. Kako and J. Ye, J. Phys. Chem. C, 2012, 116, 7621–7628 CAS.
- O. Akhavan, Carbon, 2010, 48, 509–519 CrossRef CAS.
- S. J. An, Y. Zhu, S. H. Lee, M. D. Stoller, T. Emilsson, S. Park, A. Velamakanni, J. An and R. S. Ruoff, J. Phys. Chem. Lett., 2010, 8, 1259–1263 CrossRef.
- D. Gu and J. B. Fein, Colloids Surf., A, 2015, 481, 319–327 CrossRef CAS.
- R. Sitko, E. Turek, B. Zawisza, E. Malicka, E. Talik, J. Heimann, A. Gagor, B. Feist and R. Wrzalik, Dalton Trans., 2013, 42, 5682–5688 RSC.
- J. Ran, J. Zhang, J. Yu and S. Z. Qiao, ChemSusChem, 2014, 7, 3426–3434 CrossRef CAS PubMed.
- Q. Li, H. Meng, J. Yu, W. Xiao, Y. Zheng and J. Wang, Chem.–Eur. J., 2014, 20, 1176–1185 CrossRef CAS PubMed.
- K. S. B. De Silva, S. Gambhir, X. L. Wang, X. Xu, W. X. Li, D. L. Officer, D. Wexler, G. G. Wallace and S. X. Dou, J. Mater. Chem., 2012, 22, 13941–13946 RSC.
- Y. L. Min, J. C. Fan, Q. J. Xu and S. Y. Zhang, J. Alloys Compd., 2014, 609, 46–53 CrossRef CAS.
- H. Alehdaghi, M. Marandi, M. Molaei, A. Irajizad and N. Taghavinia, J. Alloys Compd., 2014, 586, 380–384 CrossRef CAS.
- Y. Liang, M. Shao, L. Liu, J. Hu and W. Cui, Bull. Korean Chem. Soc., 2014, 35, 1182–1190 CrossRef CAS.
- J. Wang, P. Yang, J. Zhao and Z. Zhu, Appl. Surf. Sci., 2013, 282, 930–936 CrossRef CAS.
- X. Wang, H. Tian, X. Cui, W. Zheng and Y. Liu, Dalton Trans., 2014, 43, 12894–12903 RSC.
- C. M. Babu, R. Vinodh, B. Sundaravel, A. Abidov, M. M. Peng, W. S. Cha and H. T. Jang, J. Taiwan Inst. Chem. Eng., 2016, 62, 199–208 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. B. T. Nguyen and R. S. Ruof, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- G. Hota, S. B. Idage and K. C. Khilar, Colloids Surf., A, 2007, 293, 5–12 CrossRef CAS.
- C. Liangyuan, L. Zhiyong and B. Shouli, Sens. Actuators, B, 2010, 143, 620–628 CrossRef.
- J. Yan, K. Wang, Q. Liu, J. Qian, X. Dong, W. Liu and B. Qiu, RSC Adv., 2013, 3, 14451–14457 RSC.
- S. Bykkam, K. V. Rao, C. H. S. Chakra and T. Thunugunta, Int. J. Adv. Biotechnol. Res., 2013, 4, 1005–1009 CAS.
- R. Krol, Principles of Photoelectrochemical Cells, in Photoelectrochemical Hydrogen Production, ed. R. Krol and M. Gratzel, Springer, New York, USA, 2012 Search PubMed.
- Q. Mi, D. Chen, J. Hu, Z. Huang and J. Li, Chin. J. Catal., 2013, 34, 2138–2145 CrossRef CAS.
- H. Matsumoto, T. Sakata, H. Mori and H. Yoneyama, J. Phys. Chem., 1996, 100, 13781–13785 CrossRef CAS.
- A. Ye, W. Fan, Q. Zhang, W. Deng and Y. Wang, Catal. Sci. Technol., 2012, 2, 969–978 CAS.
- J. Zhong, Y. Zhang, C. Hu, R. Hou, H. Yin, H. Li and Y. Huo, J. Mater. Chem. A, 2014, 2, 19641–19647 CAS.
- N. Xu, Z. Shi, Y. Fan, J. Dong, J. Shi and M. Z. C. Hu, Ind. Eng. Chem. Res., 1999, 38, 373–379 CrossRef CAS.
- Q. Li, B. Guo, J. Yu, J. Ran, B. Zhang, H. Yan and J. R. Gong, J. Am. Chem. Soc., 2011, 133, 10878–10884 CrossRef CAS PubMed.
- Y. J. Hwang, C. Hahn, B. Liu and P. Yang, ACS Nano, 2012, 6, 5060–5069 CrossRef CAS PubMed.
- J. Zhang, W. Zhao, Y. Xu, H. Xu and B. Zhang, Int. J. Hydrogen Energy, 2014, 39, 702–710 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra04415h |
| This journal is © The Royal Society of Chemistry 2016 |