
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSPIO@SiO2–Re@PEG nanoparticles as magneto-optical dual probes and sensitizers for photodynamic therapy†
Marco
Galli
a,
Elisa
Moschini
b,
Maria Vittoria
Dozzi
a,
Paolo
Arosio
ce,
Monica
Panigati
aef,
Laura
D'Alfonso
d,
Paride
Mantecca
b,
Alessandro
Lascialfari
ce,
Giuseppe
D'Alfonso
ae and
Daniela
Maggioni
*ae
aDipartimento di Chimica, Università degli Studi di Milano, Via Golgi 19, 20133 Milano, Italy. E-mail: daniela.maggioni@unimi.it
bDipartimento di Scienze dell'Ambiente e del Territorio e di Scienze della Terra, Università degli Studi di Milano-Bicocca, Piazza della Scienza 1, 20126 Milano, Italy
cDipartimento di Fisica, Università degli Studi di Milano, Via Celoria 16, 20133 Milano, Italy
dDipartimento di Fisica, Università degli Studi di Milano-Bicocca, Piazza della Scienza 3, 20126 Milano, Italy
eConsorzio Interuniversitario Nazionale per la Scienza e Tecnologia dei Materiali, Via Golgi 19, 20133 Milano, Italy
fIstituto per lo Studio delle Macromolecole, Consiglio Nazionale delle Ricerche (ISMAC-CNR), Via E. Bassini, 15, 20133 Milano, Italy
First published on 12th April 2016
Abstract
A dual magneto-optical nanoprobe, endowed with properties useful for photodynamic therapy, has been prepared. It is constituted by a superparamagnetic iron oxide (SPIO) core (diameter size distribution centered at ca. 10 nm), obtained by thermal decomposition of iron oleate, coated by a compact silica shell, grown in a reverse microemulsion environment. Luminescent [Re(phen)(CO)3(py)]CF3SO3 complexes were covalently anchored to the silica shell, by functionalizing the pyridine ligand with a triethoxysilane moiety. Finally, the surface of the nanoparticles (NPs) was coated with a layer of polyethylene glycol (PEG), functionalized with triethoxysilane, to improve stability and stealthiness in physiological conditions. Transmission electron microscopy of these SPIO@SiO2–Re@PEG nanocomposites showed a single population, with size distribution centered at ca. 40 nm. NPs showed nuclear relaxivity values that guarantee an appreciable contrast in magnetic resonance imaging (r2 > 30 s−1 mM−1 at frequencies higher than 5 MHz). The presence of the Re complexes imparted photoluminescence to the NPs, with blue shifted emission and higher photoluminescence quantum yields with respect to the free [Re(phen)(CO)3(py-upts)]+ complex (λem 553 vs. 580 nm, Φ 0.060 vs. 0.038, in aerated water solution). The complexes embedded into the NPs maintained a satisfactory efficiency toward 1O2 generation (quantum yields 0.21 vs. 0.26 for the free complex, as assessed using 1,5-dihydroxynaphthalene as indirect marker of the 1O2 presence). Preliminary cell penetration tests were performed on human lung adenocarcinoma A549 cells. Two photon excitation confocal microscopy showed that the NPs were easily internalized and accumulated in the perinuclear region of the cells already after 4 h of incubation. Increased cytotoxicity upon irradiation with respect to the dark was also observed, showing the potential of the nanocomposite for photodynamic therapy applications.
1. Introduction
The use of nanoparticles (NPs) in biomedicine has attracted much attention in the past decades, for their remarkable potential in the resolution of many long-standing problems.1 For this purpose, the NPs diameters must be in the range 10–100 nm, since smaller particles would escape from the circulatory system through renal clearance, whereas larger ones will be recognized by reticulo-endothelial system (RES), sequestered and mainly delivered to the liver. Preferential accumulation in solid tumors can be observed for NPs with size smaller than 100 nm, for the so called EPR (enhanced permeability and retention) effect, which can be further enhanced if the surface is functionalized with molecules or polymers able to make the NPs stealth to the RES.2,3 NPs, thanks to their large surface area, can be loaded with huge amounts of small molecules, and therefore can act as efficient carriers of drugs or imaging agents toward their target, preventing their rapid clearance. In this way different diagnostic and therapeutic functionalities can be integrated in a single nanosized system. Since each imaging technique has pros and cons, nanoprobes provided with different contrast agents exploitable in different imaging techniques could provide many advantages over single monomodal probes.Many dual magneto-optical probes have already been reported,4–8 in which iron oxides magnetic NPs bear light emitters, like quantum dots (QDs)9 or organic fluorophores.10 However the high toxicity of QDs limits their application in diagnosis, while organic fluorescent molecules often present drawbacks, such as high photobleaching, short lifetimes, and emission energy close to cell autofluorescence. The use of organometallic complexes emitting from triplet states can overcome these disadvantages, due to their generally high photostability, large Stock's shifts, which prevent self-quenching and overlapping with cell autofluorescence, and long lifetimes, which allow to improve the sensitivity by the use of time-resolved techniques. For these reasons phosphorescent metal complexes have been often proposed for imaging techniques.11
Among these phosphorescent emitters, diimine complexes of fac-Re(CO)3+ have been attracting much interest since long time, for their remarkable photophysical properties,12 which have been exploited in a variety of fields, either for biomedical applications,13 or material science.14 Like other d6 transition-metal complexes, these species exhibit an intense long-lived emission, originating from triplet metal-to-ligand charge transfer (3MLCT) excited states. The strongest emissions occur either from mononuclear cationic complexes15,16 or from dinuclear species in which two metal centers are bonded to the same chromophore.17 The direct conjugation of a luminescent mononuclear rhenium complex “Re(L)(CO)3+” to magnetite NPs has been recently reported,18 and this nanosystem is proposed as multimodal probe MRI T2 contrast agent, optical probe, and potential β and γ emitter through the use of 186/188Re hot isotopes, for single-photon emission tomography (γ) or radiotherapy (β).
It has been recently proven that some of these rhenium complexes are endowed with another important property for biomedical uses, i.e. they are efficient sensitizers for the generation of singlet oxygen,19–23 by triplet–triplet energy transfer. Singlet oxygen production is at the base of photodynamic therapy (PDT),24–27 which has become more and more attractive as a valid coadjuvant (or even alternative) to chemotherapy in cancer cure and as a new method for killing pathogens in localized infections.
PDT requires a photosensitizer, i.e. a molecule that can be photo-excited in a triplet state and is able to transfer energy to molecular oxygen, converting it from triplet to singlet state (eqn (1)).
| 3S*1 + 3O2 → 1S0 + 1O2 | (1) |
Among the various species used as photosensitizers we can list organic dyes as methylene blue28 or rose Bengal,29 as well as porphyrins,24 chlorines,26,30 phthalocyanines,24 porphycenes,31,32 and metal complexes of Ru,24 Ir,33–35 Pt36,37 and Re.19–23 Many different nanomaterials for PDT containing transition metal complexes as photosensitizers have also recently appeared in the literature,38–42 One only of the above nanosized systems, constituted by silica NPs, involved the use of rhenium complexes.42
In this work we have prepared a dual probe nanocomposite, constituted by an iron oxide magnetic core and a compact silica shell functionalized with a luminescent complex, viz. [Re(phen)(CO)3(py-upts)]CF3SO3 (py-upts = 1-(pyridin-4-yl)-3-(3-(triethoxysilyl)propyl)urea). The silica shell has been coated by a polyethyleneglycol (PEG) layer. Actually, pegylation, though known to partly affect the efficiency of nanoparticles uptake, is a very effective method to reduce their toxicity.43 Moreover, polyethylene glycols in vitro enhance the nanoparticles stability and in vivo allow the nanoparticles to avoid macrophage recognition, uptake and clearance from systemic circulation.44 The photophysical and relaxivity properties of the SPIO@SiO2–Re@PEG nanocomposites have been measured, to check the possible mutual interference between the magnetic core and the luminescent shell. Preliminary cell penetration tests have been conducted on human lung adenocarcinoma A549 cells to investigate the internalization of SPIO@SiO2–Re@PEG NPs as well as the molecular probe, by means of two photon excitation (TPE) confocal microscopy. The ability of the nanocomposites to act as PDT agents has also been investigated, both in cuvette and in vitro.
2. Experimental
Materials, Instruments and Methods are detailed in ESI,† as well as the synthesis of the SPIO@OA NPs, of the py-upts ligand and of PEG500-silane.2.1. Synthesis of the [Re(phen)(CO)3(py-upts)]OTf complex
Re(CO)5OTf45 (OTf = CF3SO3, 37.2 mg, 7.83 × 10−2 mmol) was dissolved in dry toluene (8 mL) under nitrogen, and added with 15.5 mg of 1,10-phenanthroline monohydrate (7.8 × 10−2 mmol). The mixture was heated at 100 °C for 5 h. Finally, the temperature was lowered to 0 °C, giving an orange powder, from which the intermediate product [Re(phen)(CO)3(OTf)] was isolated by washing with toluene (3 mL, 2 times) and drying under vacuum. Yield 96%. The progress of the reaction was monitored by IR spectroscopy and the final IR spectrum showed ν(CO) bands (CH2Cl2) at 2035 (vs.), 1935 (s) and 1915 (s) cm−1. The pyridine ligand py-upts (20.9 mg, 0.0644 mmol) was dissolved in 1 mL of anhydrous ethanol under nitrogen. Then, a sample of [Re(phen)(CO)3(OTf)] (38.6 mg, 0.0643 mmol), dissolved in 2 mL of EtOH, was added. The initial suspension was heated at 50 °C and left to react under magnetic stirring and nitrogen for 48 h, obtaining a clear and yellow solution. In order to avoid the hydrolysis of the ethoxy groups, the resulting complex [Re(phen)(CO)3(py-upts)]OTf was not isolated and was stored at 4 °C in ethanol solution under nitrogen. The completeness of the coordination of py-upts was assessed by 1H-NMR. 1H NMR (400 MHz, MeOD) δ 9.72 (dt, J = 5.1, 1.2 Hz, 2H), 8.95 (dt, J = 8.3, 1.2 Hz, 2H), 8.25 (s, 2H), 8.13 (dd, J = 8.3, 5.1 Hz, 2H), 8.05 (d, J = 6.0 Hz, 2H), 7.19 (d, J = 6.0 Hz, 2H), 3.79 (quart, J = 7.2 Hz, 6H), 3.11 (m, 2H), 1.53 (m, 2H), 1.18 (t, J = 7.2 Hz, 9H), 0.57 (m, 2H). FTIR ν(CO): 2034 (vs.), 1934 (br, s) cm−1 (ethanol); 2033 (vs.), 1924 (br, s) cm−1 (water).2.2. Coating of the SPIO NPs with a SiO2 shell
The silica coating was obtained by a synthesis conducted in a reverse microemulsion. IGEPAL CO-520 (2.7385 g, 6.21 mmol) was dissolved by sonication (20 min) in 22 mL of cyclohexane, in a Schlenk round bottom flask. Then SPIO@OA NPs (0.82 mg, see ESI†) were added, together with aqueous NH3 (28%, 200 μL) and TEOS (150 μL, 0.677 mmol) to give a brown clear solution. The microemulsion was stirred for 16 h at room temperature, then treated with ethanol (20 mL) and centrifuged (7550 g, 20 min). The precipitate was recovered and easily re-suspended in 20 mL of ethanol by sonication. The purification procedure was repeated twice.2.3. Functionalization of the SPIO@SiO2 NPs with [Re(phen)(CO)3(py-upts)]OTf and PEG500-silane
The ethanol suspension of SPIO@SiO2 NPs was treated with an ethanol solution of the complex [Re(phen)(CO)3(py-upts)]OTf (310 μL, containing ca. 1 mg of complex, see ESI†) and a few drops of aqueous NH3 (28%), as catalyst. The suspension was refluxed for 5 h, by magnetic stirring under nitrogen, then the mixture was cooled to room temperature, and treated with H2O (4 mL), aqueous NH3 (28%, 600 μL) and PEG500-silane (60 μL, ca. 0.126 mmol, see ESI†). The reaction was allowed to proceed under magnetic stirring for 48 h. Then, the NPs were isolated by centrifugation (15 min at 7550 g) and finally suspended in 20 mL of milliQ water and stored at 4 °C for further uses. FTIR (water) ν(CO): 2033 (vs.), 1924 (br, s) cm−1. The content of Fe and Re in the mother solution was determined by ICP-AES on a sample digested as follows: 1.00 mL of the NP suspension was dried on a sand bath, added with 1 mL of a 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 mixture of concentrated HCl and HF (36% and 48%, respectively), and allowed to react for 30 min under sonication in a Teflon vial at room temperature. The excess of HF was quenched by addition of H3BO3 (2 mmol). Then, a mixture of concentrated HNO3 and H2O2 (1:2 ratio, 1.5 mL) was added and left to react overnight at room temperature. [Fe] from ICP-AES = 1.2 × 10−4 M (6.7 μg mL−1 Fe); [Re] from ICP-AES = 3.8 × 10−5 M.
:30 mixture of concentrated HCl and HF (36% and 48%, respectively), and allowed to react for 30 min under sonication in a Teflon vial at room temperature. The excess of HF was quenched by addition of H3BO3 (2 mmol). Then, a mixture of concentrated HNO3 and H2O2 (1:2 ratio, 1.5 mL) was added and left to react overnight at room temperature. [Fe] from ICP-AES = 1.2 × 10−4 M (6.7 μg mL−1 Fe); [Re] from ICP-AES = 3.8 × 10−5 M.
2.4. Photochemical stability test on [Re(phen)(CO)3(py-upts)]OTf
A sample of [Re(phen)(CO)3(py-upts)]OTf was dissolved in water saturated with O2. The solution was exposed to visible light (by using a 150 W/NDL lamp, 390 nm cutoff filter, 142 mW cm−2 see Fig. S1†), and UV-vis absorption spectra were recorded every 15 min for 1 h (Fig. S2†).2.5. Singlet oxygen production
In a quartz cuvette 0.555 mL of the above described mother solution of SPIO@SiO2–Re@PEG NPs were added to 1.70 mL of a H2O/MeOH 80:20 mixture, affording a [Re] = 1 × 10−5 M. Subsequently 1,5-dihydroxynaphthalene (DHN) (0.25 mL of a MeOH solution 1 × 10−3 M) was added to give a [DHN] = 1 × 10−4 M. Oxygen was bubbled in the cuvette for 10 min. The solution was then irradiated with visible light (λ > 390 nm) and the reaction was followed by acquiring UV-vis absorption spectra at different times. The same procedure was followed for the photoreactions involving other photosensitizers, i.e. [Re(phen)(CO)3(py)]OTf (1 × 10−5 M), and methylene blue (MB, 8 × 10−7 M, the much lower concentration being necessary to balance the much higher molar absorptivity of MB).
2.6. Cell uptake and cytotoxicity in dark and light conditions
SPIO@SiO2–Re@PEG-NPs were sonicated 30 minutes at 40 kHz (Sonica – Soltec) just before the use in order to obtain a homogenous dispersion and then different volumes of the stock suspension were added directly in the culture medium to obtain the working concentrations. The alveolar epithelial cells A549 from human lung carcinoma, purchased from ECACC (European Collection of Cell Cultures), were routinely maintained in OptiMEM 10% FBS, at 37 °C, 5% CO2 and seeded in 6 multi-well plates for the cell uptake assays. Images of treated-cells were recorded after exposure for 4 h or 24 h to: (a) 50 μL of a 760 μg mL−1 suspension of NPs affording a NP concentration of 19 μg mL−1, corresponding to a [Re] = 0.95 μM in the well; (b) 50 μL of a water solution (3.2 × 10−4 M) of the precursor molecule [Re(phen)(CO)3(py-upts)]OTf, affording a final [Re] = 8.0 μM in the well. Two photon excitation at 840 nm was exploited (with an excitation power on the sample of about 15 mW).The A549 cells were also seeded in 12 multi-well plates for cell viability evaluation. Cells were exposed to increasing concentrations of NPs (1.9; 9.5; 19; 38; 76 μg mL−1) and then incubated for 4 h at 37 °C, 5% CO2 to allow NP internalization. At the end of this pre-incubation period the A549 cells were irradiated with the 150 W/NDL lamp light (200 mW cm−2) for 10 min in order to photoactivate the internalized nanostructured compound and then they were incubated again for 3 h or 16 h. Not irradiated cells exposed at the same concentrations of NPs were used as reference (dark conditions). At the end of the exposure, cells were rinsed with PBS and the working MTT solution prepared in culture medium at a concentration of 0.3 mg mL−1 was added. After 2 h the MTT solution was removed and the purple MTT reduction product (formazan salts) was solubilized with DMSO. The optical density of each sample, proportional to cell viability, was measured at 570 nm using 690 nm as reference wavelength with a multiplate reader. The experiments were replicated three times and results were expressed as mean ± SE. Statistical differences were verified using One way ANOVA followed by Fischer LSD test (p < 0.05).
3. Results and discussion
3.1. Synthesis of the complex [Re(phen)(CO)3(py-upts)]OTf
As discussed in the Introduction, a [Re(phen)(CO)3(py)]+ complex has been chosen as luminescent probe and singlet oxygen photosensitizer. In order to avoid its slow release from the inorganic matrix, the pyridine ligand was functionalized with a triethoxysilane moiety, to covalently bind the complex to the silica shell. The ligand 1-(pyridin-4-yl)-3-(3-(triethoxysilyl)propyl)urea,46 hereafter py-upts, was obtained as depicted in Scheme 1, using a slightly modified literature procedure.47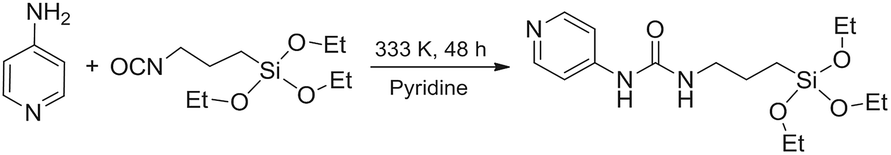 | ||
| Scheme 1 Reaction scheme for the synthesis of the py-upts ligand. | ||
Then the [Re(phen)(CO)3(py-upts)]OTf complex was prepared, by replacing with py-upts the labile triflate ligand in the precursor [Re(phen)(CO)3(OTf)] complex (Scheme 2).48 The reaction was carried out in anhydrous ethanol, to avoid the early hydrolysis of the three ethoxy groups of the silane moiety, and its progress was monitored by 1H NMR (Fig. S3†). The entry of pyridine in the ligand sphere was accompanied by the progressive shift of the photoluminescence, from a faded orange to a brilliant yellow color.
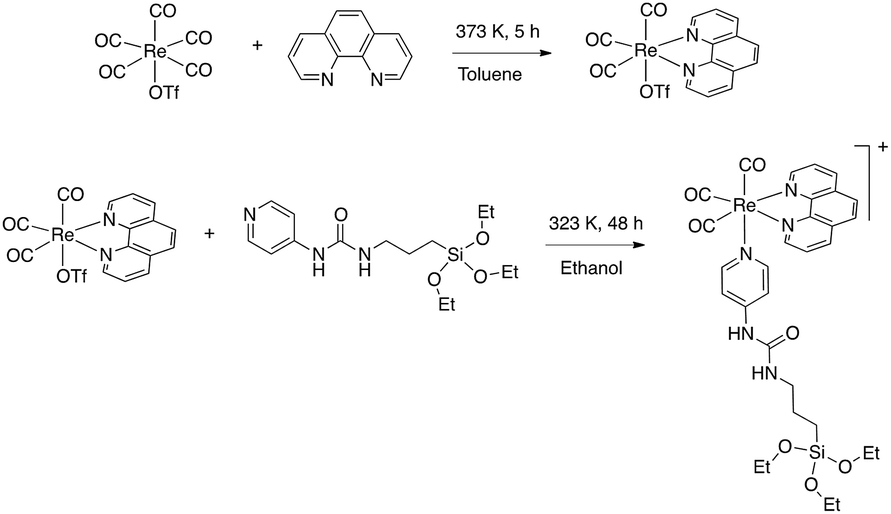 | ||
| Scheme 2 Synthetic route to the complex [Re(phen)(CO)3(py-upts)]OTf. | ||
3.2. Synthesis of the magneto-optical SPIO@SiO2–Re@PEG nanocomposites
The synthesis of iron oxide NPs stabilized by an oleic acid shell was performed by a literature procedure,50 involving thermal decomposition of iron oleate in a high boiling solvent (here 1-octadecene). NPs with a diameter size distribution centered at 10.4 ± 1.3 nm (from DLS analysis, Fig. S4†) were obtained. The transmission electron microscopy (TEM) images (Fig. 1) were in good agreement with the DLS data, showing a size distribution of the magnetic core centred at 9.4 ± 1.3 nm. | ||
| Fig. 1 TEM micrograph of the SPIO@OA NPs (up) and of the core/shell SPIO@SiO2–Re@PEG NPs (down), with the corresponding size distribution histograms. | ||
The silica shell was grown in a reverse microemulsion environment,51,52 and the reaction was stopped after 6 h, when DLS indicated the prevalence of NPs with a hydrodynamic diameter of about 45 nm (Fig. S5†).
The luminescent [Re(phen)(CO)3(py-upts)]OTf complexes were then anchored to the silica shell, by treating the SPIO@SiO2 NPs, dispersed in ethanol, with an ethanol solution of the complex, as depicted in the second step of Scheme 3. The mixture was refluxed for 5 h, and the precipitate was recovered by centrifugation (see Experimental part). The solid was strongly luminescent and was also attracted by an external permanent magnet (Fig. S6†), thus immediately indicating the formation of a dual magneto-luminescent nano-system (as subsequently demonstrated by the detailed characterization described in the following sections).
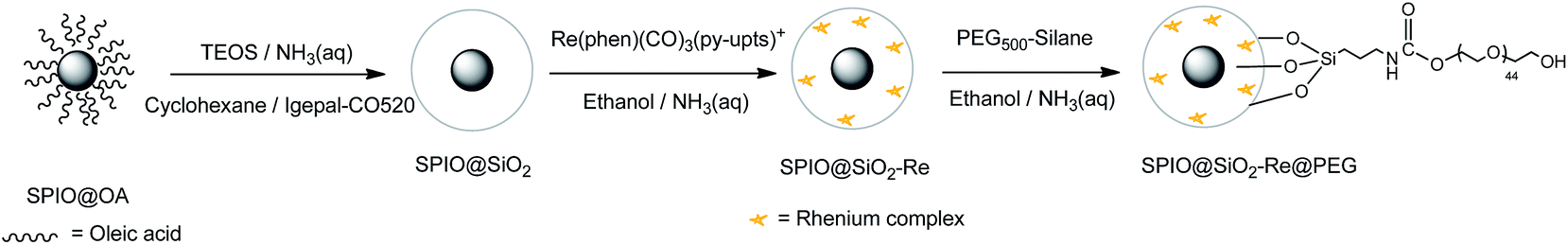 | ||
| Scheme 3 Schematic steps diagram of the synthesis of PEG-capped magneto-luminescent SPIO@SiO2–Re@PEG NPs. | ||
Attempts to insert the luminescent probe directly in the water-in-oil microemulsion, to condense its silane terminals to the SPIO@SiO2 inside the nanoreactor, failed, since significant aggregation was observed, likely due to destabilization of the microemulsion after the treatment with the ethanol solution of the Re complex.
Upon water addition the SPIO@SiO2-Re NPs were easily suspended, but the stability of the colloidal dispersion was poor and in few hours the NPs completely settled. For this reason, and also to make them stealth to the reticulo-endothelial system for any in vivo future use, the NPs were stabilized by capping their surface with short polyethylene glycol molecules (last step in Scheme 3), previously functionalized with a triethoxysilane group through a carbamate link (Scheme 4 and Fig. S7†).53 After the coating with the PEG500-silane, the ζ potential of the NPs changed from −20 mV to 0 mV and the hydrodynamic diameter increased from 45 nm to about 80 nm (Fig. S8†). Moreover, the thermogravimetric analysis of the nanocomposite (Fig. S9†) revealed an overall loss weight of about 60%, ascribable to PEG and to the ligands on the rhenium complex.
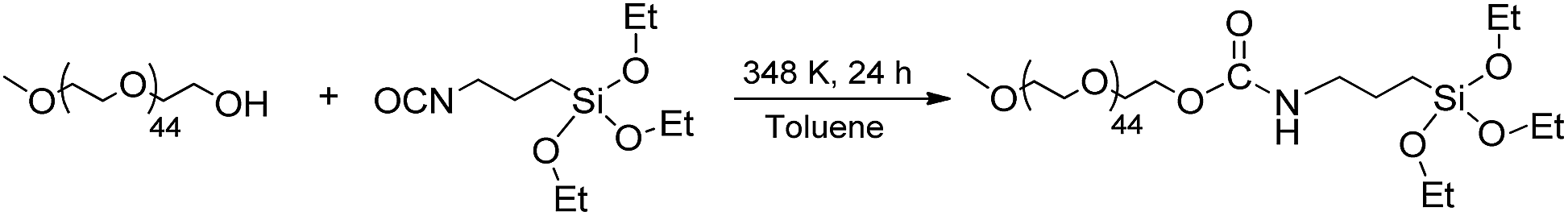 | ||
| Scheme 4 The preparation of the PEG silanized by formation of a carbamate link. | ||
The presence of the polymer shell strongly improved the stability of the colloid and reduced the formation of irreversibly aggregated NPs.
The morphology of the magneto-luminescent SPIO@SiO2–Re@PEG nanocomposite was analyzed by TEM (Fig. 1), which revealed a spherical form and a single magnetic core per core/shell nanocomposite. The size distribution of the nanocomposite showed a single population centered at 40 ± 3.5 nm.
The amount of Re on the NPs was estimated by ICP-AES analysis, corresponding to a loading of ca. 6000 Re atoms per NPs. This loading well compares with the one obtained for related Ru luminescent complexes covalently embedded into silica NPs with diameters of ca. 60 nm (4600 Ru atoms per NP).54
3.3. Photoluminescence properties of the precursor Re complex and of the SPIO@SiO2–Re@PEG NPs
The photophysical characterization of the free complex [Re(phen)(CO)3(py-upts)]OTf and of the corresponding SPIO@SiO2–Re@PEG nanocomposite was performed in dilute, air equilibrated, water solution (about 1.0 × 10−5 M), as well as in the solid state. In order to evaluate the influence of the urea link in the para position of the pyridine ligand, the photophysical properties of the precursor complex were compared with those of the already known [Re(phen)(CO)3(py)]OTf complex16 containing the unsubstituted pyridine ligand. The most relevant photophysical data are listed in Table 1 and the corresponding absorption and emission spectra are displayed in Fig. 2.| Compound | Water solution, air | Solid | ||||
|---|---|---|---|---|---|---|
| λ em/nm | τ/ns | Φ | λ em/nm | τ/ns | Φ | |
| [Re(phen)(CO)3(py)]OTf | 567 | 454 | 0.070 | 530 | 740 (46%) | 0.158 |
| 2570 (54%) | ||||||
| [Re(phen)(CO)3(py-upts)]OTf | 580 | 257 | 0.038 | 553 | 993 (30%) | 0.119 |
| 2032 (70%) | ||||||
| SPIO@SiO2–Re@PEG NPs | 553 | 409 (22%) | 0.060 | |||
| 1738 (78%) | ||||||
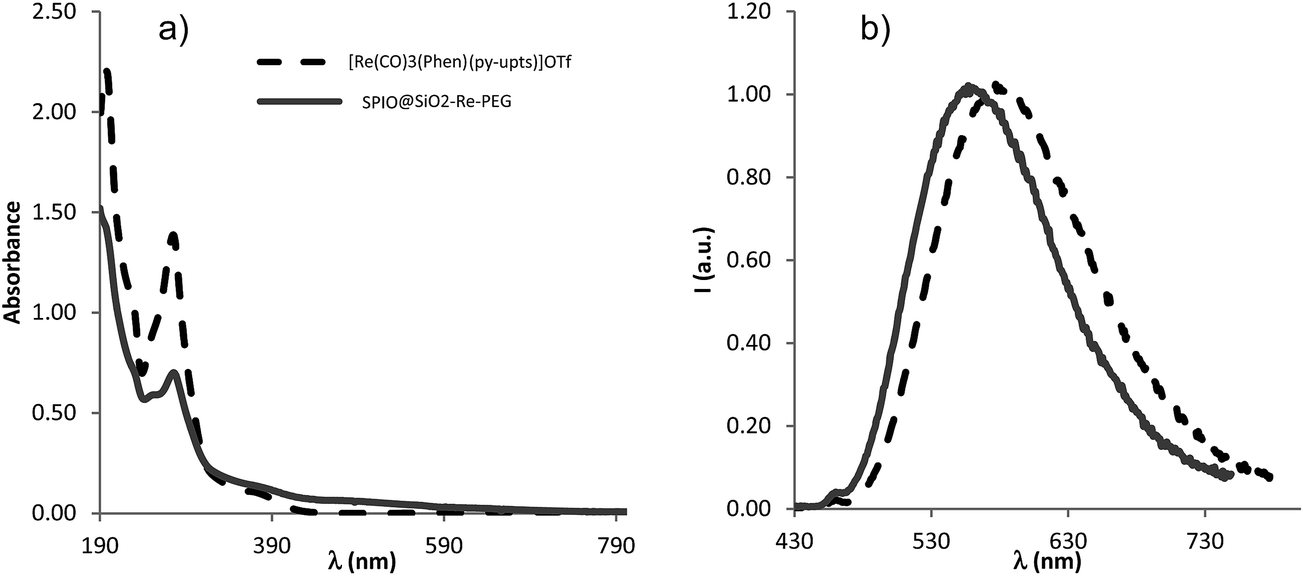 | ||
| Fig. 2 (a) UV-vis absorption and (b) normalized photoluminescence spectra (λex = 375 nm) of [Re(phen)(CO)3(py-upts)]OTf (black dashed traces) and SPIO@SiO2–Re@PEG NPs (gray traces) in air equilibrated water solution at room temperature. | ||
The UV-vis absorption spectra of the free complexes and the NPs showed a strong band at about 275 nm, ascribed to ligand-centered (LC) π–π* transitions, and a weaker broad band, centered at about 370 nm. The latter band is ascribable to a spin allowed dπ(Re) → π*(phen) metal-to-ligand charge transfer transition (1MLCT), by comparison with analogous cationic Re(I) tricarbonyl complexes.15,16
Upon optical excitation the free complexes displayed a broad and featureless emission band in the green-yellow region of the visible spectrum, arising from an excited state that can be described as a triplet MLCT state.12 This was confirmed by the quenching of the emission on going from de-aerated to air equilibrated solution (yields dropping from 0.117 to 0.070 for [Re(phen)(CO)3(py)]OTf, and from 0.053 to 0.038 for [Re(phen)(CO)3(py-upts)]OTf) and by the solvent-dependence of the emission maximum, which results red-shifted by about 20 nm on going from CH2Cl2 (λ = 544 nm)16 to water (λ = 567 nm, Table 1) for the complex [Re(phen)(CO)3(py)]OTf.
The MLCT nature of the excited state was further supported by the influence of the pyridine substituents on the absorption and emission spectra. Actually, the introduction of the electron-donating (by resonance effect) urea link in the para position of the pyridine ligand, afforded a red-shift of about 13 nm of the emission maximum of [Re(phen)(CO)3(py-upts)]OTf with respect to [Re(phen)(CO)3(py)]OTf. This is in line with the presence of the electron-richer py-upts ancillary ligand,55 which raises the HOMO level, then lowering the energy of the MLCT transition. The red shift of the emission energy, observed for the complex [Re(phen)(CO)3(py-upts)]OTf, was accompanied by a slight decrease of the photoluminescence quantum yield (Φ) and of the lifetime of the excited state (Table 1), in agreement with the energy gap law.56
The photophysical properties of the complexes were investigated also in the solid state, showing a blue shift of the emission maxima with respect to the solution, in agreement with the rigidochromic effect usually observed for mononuclear rhenium complexes.57 An increase of the Φ values was also observed, indicating that here the quenching processes often observed in the solid state do not occur, and the dominant effect is the decrease, in the rigid environment, of the roto-vibrational motions, responsible for the non-radiative deactivation pathways of the excited states.17a,c
Some interesting differences in the photophysical behavior were observed on going from the molecular complexes to the NPs. The emission maximum of the NPs in water solution was considerably blue-shifted with respect to the corresponding [Re(phen)(CO)3(py-upts)]+ complex in the same conditions, to a value very similar to the one measured for this complex in the solid state. Moreover, for the NPs the excited state lifetimes were longer and the emission quantum yields higher than the values measured for the corresponding py-upts complex in solution. This can be attributed both to the confinement of the complex in the rigid silica environment and to its poor exposition to the solution, hampering the deactivating action of water. However, the complex in the (porous) silica shell remained sensitive to oxygen presence, as indicated by its ability to generate singlet oxygen (see paragraph 3.6).
The two excited state lifetimes observed for the NPs, as well as for the complexes in the solid state, result from the presence of complexes experiencing different environments, with different available non-radiative deactivation pathways.
3.4. Relaxometry properties of SPIO@SiO2–Re@PEG NPs
The 1H NMR longitudinal and transverse relaxation times of the SPIO@SiO2–Re@PEG NPs were measured at room temperature in the frequency range to cover most of the typical fields for MRI tomographs, used both in clinics (H = 0.2, 0.5, and 1.5 T) and in research laboratories,58 as specified in the experimental part. The efficiency of the contrast agents was evaluated in the usual way: the nuclear relaxivities, both r1 and r2, were calculated as the inverse of the relaxation times normalized for contrast agent concentration, i.e. the concentration of the magnetic part, once previously corrected for the host diamagnetic contribution, according to eqn (2).| ri = [(1/Ti)meas − (1/Ti)dia]/c, i = 1, 2 | (2) |
Fig. 3 shows the r1 and r2 nuclear relaxivities of SPIO@SiO2–Re@PEG NPs together with the values for the commercial contrast agent Endorem®. The displayed r2 values for υ > 5 MHz (r2 > 30 s−1 mM−1) guarantee an appreciable contrast in the MR images once the nanoparticles are used, as some of us already demonstrated on other T1 and/or T2 relaxing nanoparticles of completely different types.59
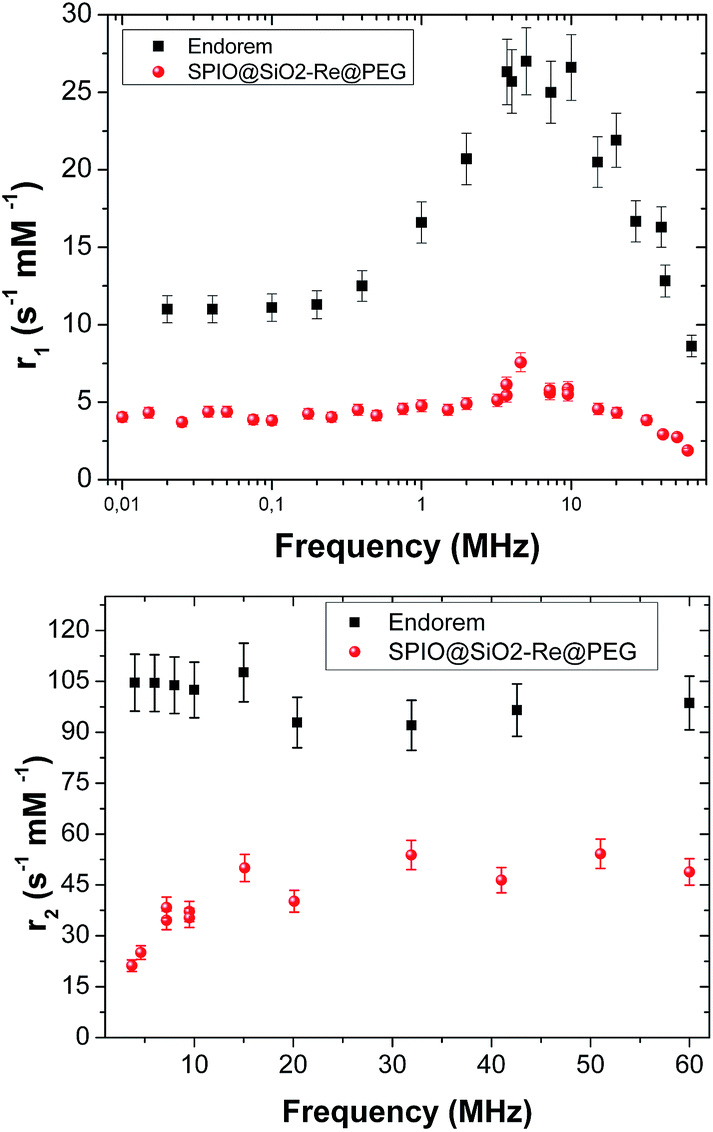 | ||
| Fig. 3 Longitudinal (top) and transverse (bottom) relaxivities of SPIO@SiO2–Re@PEG NPs (red circles) dispersed in water. Data are compared with the commercial compound Endorem® values (black squares). | ||
3.5. Singlet oxygen generation by SPIO@SiO2–Re@PEG NPs
The photochemical stability of the photosensitizer was preliminarily tested, by irradiating for 1 h a sample of the free complex [Re(phen)(CO)3(py-upts)]OTf, dissolved in water saturated with O2 (Fig. S2†). The superposition of the absorption spectra recorded at different times indicated the high photostability of the photosensitizer in these conditions.The efficiency of the SPIO@SiO2–Re@PEG NPs toward singlet oxygen generation was assessed by using 1,5-dihydroxynaphthalene (DHN) as indirect marker of the 1O2 presence. Actually, DHN reacts quantitatively with 1O2 to give the oxidized species juglone (5-hydroxy-1,4-naphtalenedione, Scheme 5), according to eqn (3).35,60,61
| DHN + 1O2 → juglone + ½3O2 | (3) |
 | ||
| Scheme 5 Photochemical oxidation of DHN to juglone in the presence of a photosensitizer (PS). | ||
Thus, the 1O2 production was indirectly monitored by following the decrease of the UV-vis absorption band of DHN at 297 nm (accompanied by the increase of the juglone band at 427 nm). Fig. 4A shows the spectra recorded for the first 60 min of irradiation of a sample of NPs suspended in a MeOH/H2O (80:20) mixture, in the presence of DHN.
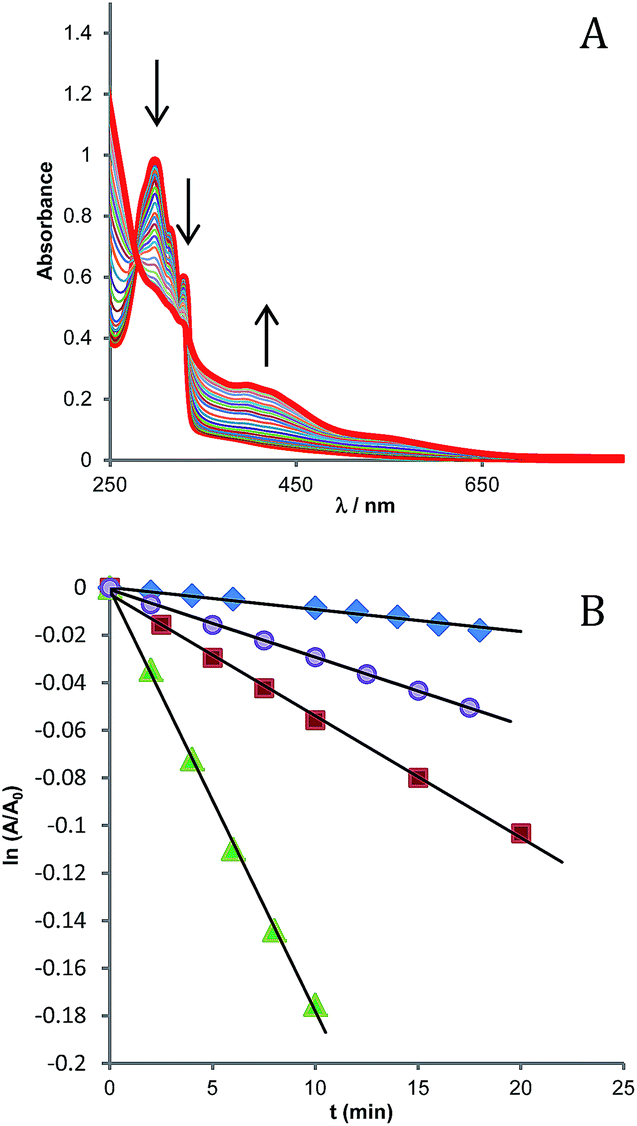 | ||
| Fig. 4 (A) UV-vis absorption spectra recorded at different irradiation times (from 0 to 360 min, λ > 390 nm) on a solution containing SPIO@SiO2–Re@PEG NPs (150 μg mL−1) and DHN (10−4 M) in 2.5 mL of MeOH/H2O (80/20) bubbled with O2 for 10 min. (B) Photo-oxidation of DHN in the presence of different sensitizers: methylene blue (△), [Re(phen)(CO)3(py)]OTf (O), [Re(phen)(CO)3(py-upts)]OTf (□), and SPIO@SiO2–Re@PEG NPs (◊). At and A0 represent the absorbance measured at 297 nm (the maximum of the DHN absorption band) at time t and time 0, respectively. | ||
The reaction in the initial stages follows a pseudo-first order kinetics, ν = kobs[DHN], provided that the oxygen concentration within this interval of time can be considered constant.61 By plotting as a function of the irradiation time the values of ln(A/A0) for the DHN absorption at 297 nm (Fig. 4B), the kinetic constant kobs for reaction 3 can be estimated.
The reaction was also carried out employing as photosensitizers the precursor complex [Re(phen)(CO)3(py-upts)]OTf, or the analogue [Re(phen)(CO)3(py)]OTf complex (for comparison), and methylene blue (MB) as standard.
The quantum yields for singlet oxygen generation (ΦΔ) were determined using eqn (4):
| ΦΔ = ΦstdΔ × (νiIstd/νstdiI) | (4) |
It must be pointed out that the ΦΔ values estimated for dyes entrapped into nano-supports are affected by significant uncertainties, because of the extinction arising from light scattering (which varies with wavelength and can be only partly corrected)63 and also because of many other possible matrix effects (including here the absorption of the iron oxide core), which are difficult to accurately evaluate.64 All these contributions to the absorption spectra do not contribute to the excitation of the sensitizer, and therefore their inclusion in the computation of the I value can lead to severe underestimation of the quantum yields. For these reasons we preferred to obtain an approximate value of the photons absorbed by the rhenium dye in the NPs by using the absorption spectrum of the free complex, corrected for the different concentrations in the two solutions (that were known from the ICP analysis of the rhenium content).
The ΦΔ values estimated in this way for the SPIO@SiO2–Re@PEG NPs resulted very similar to the ones evaluated for the free Re complexes (0.26–0.29, see Table 2), which well compare with those reported for analogous tricarbonyl Re complexes (0.20–0.26) in water solution.20,65 Due to the approximations involved in the evaluation of the light absorbed by the Re complexes on the NPs, we cannot rule out that the actual quantum yields were somewhat lower than reported in Table 2. In fact some decrease of the ΦΔ value might result from the fact that 3O2 has to diffuse across the NP coating in order to quench the triplet excited state of the anchored dyes and from the possibility of entrapment of generated 1O2 in the NP matrix. However, the significant point is that the NPs do maintain a significant sensitizing activity. A lowest limit to this activity (ΦΔ = 0.05) was computed using the unprocessed absorption spectra of the SPIO@SiO2–Re@PEG NPs, i.e. the spectrum including all the ineffective contributions.
| k obs × 103 (min−1) | I (mW cm−1) | Φ Δ | |
|---|---|---|---|
| Methylene blue | 17.8 | 4.63 | 0.50 |
| [Re(phen)(CO)3(py-upts)]OTf | 5.30 | 2.64 | 0.26 |
| [Re(phen)(CO)3(py)]OTf | 2.91 | 1.31 | 0.29 |
| SPIO@SiO2–Re@PEG NPs | 0.925 | (0.57) | (0.21) |
This is remarkably different from what observed for a very effective sensitizer as MB, for which a strong decrease in the 1O2 release (about two order of magnitude) when entrapped in a silica layer was measured in a previous work.67 This might be due to some triplet–triplet annihilation process, arising from the close spatial confinement of a large number of emitters with very long excited state lifetimes, as it is typical of the triplet states of organic dyes. On the contrary, for the rhenium complex, which has a relatively short lifetime of the triplet state, the efficiency in singlet oxygen generation was maintained.
3.6. Cell uptake and PDT cytotoxicity of the SPIO@SiO2–Re@PEG nanoparticles
The cell uptake of the nanocomposites was preliminarily investigated, to find the time scale useful for the PDT assays. The uptake by A549 cells was monitored by confocal microscopy, exploiting the photoluminescence of the Re complexes, and in particular the possibility to promote their emission by two-photon excitation (TPE). TPE microscopy (as described first by Denk et al.)68 uses near-infrared laser light to excite chromophores with a (non-linear) quadratic dependence of the excitation probability from the laser power, inducing optical sectioning without the need of a pinhole (spatial filter). It presents many advantages for in vivo applications: the near-infrared light displays a deeper penetration in highly scattering tissues, and it induces a much lower photodamage of the sample with respect to UV radiation. The intrinsic optical sectioning, moreover, limits the possible phototoxic effect to the focal plane only, and increases the detection efficiency of the whole system, since it does not require descanning optics nor pinholes in the revelation path, allowing the use of large active area detectors.Cells were incubated either with the molecular probe [Re(phen)(CO)3(py-upts)]OTf or with the SPIO@SiO2–Re@PEG NPs, at different concentrations (see Experimental), and observed after both 4 h and 24 h through TPE confocal microscopy. Nuclei were stained with the viable nuclear dye Hoechst (blue), just before the microscopy evaluation.
The internalization of the free Re complex was hampered by its low solubility in aqueous media, which led to the formation of microsized aggregates, well visible in the images acquired after 4 h of incubation (Fig. S10A†). However, a moderate cell uptake was observed at longer times (24 h, Fig. S10B†), and the complexes accumulated in the cytoplasm, forming small endosomal-like clusters.
On the contrary, the nanoparticles were more easily internalized and accumulated in the perinuclear region of the cells already after 4 h of incubation (see Fig. 5B and the movie of a z-scan images sequence, downloadable from ESI†), although their persistence in the cytoplasm at longer times was not confirmed. The feeble luminescence observed after 24 h (Fig. 5C) was mainly localized outside the cell membranes, suggesting that most of the probes had been excreted by the cells. On the bases of these results an incubation time of 4 h was set for the cytotoxicity assays.
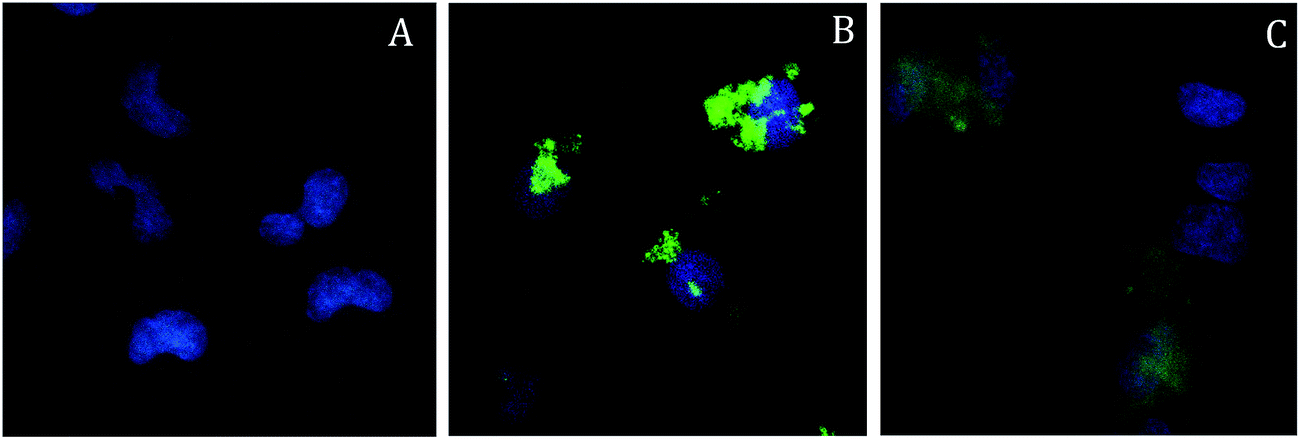 | ||
| Fig. 5 TPE microscopy images, result of the projection and superposition on the xy plane of 10 single planes acquired along the z axis at 0.5 μm steps, of A549 cells incubated with SPIO@SiO2–Re@PEG NPs (19 μg mL−1, corresponding to [Re] = 0.95 μM) for 4 h (panel B) and 24 h (panel C). Panel A shows untreated A549 cells used as control for the same experiment. The blue color is due to the staining of nuclei with Hoechst. | ||
The cytotoxic effect of the SPIO@SiO2–Re@PEG NPs was assessed both in dark and light conditions, to investigate the ability of the NPs to induce an increase in cell death when photo-activated. Cells were incubated with different concentrations of NPs for 4 h, then were either irradiated for 10 min with light from a 150 W NDL lamp or kept in the dark for the same time, and finally incubated again for 3 h or 16 h. At the end, the cell viability was evaluated by the MTT test (see Experimental part).
The results are summarized in Fig. 6. After 3 h and 16 h post irradiation (p.i.), a dose dependent effect induced by the NPs was observed, with a statistically significant increase in cell mortality after irradiation of the cells, compared to the dark exposure, testifying for an effective photodynamic response.
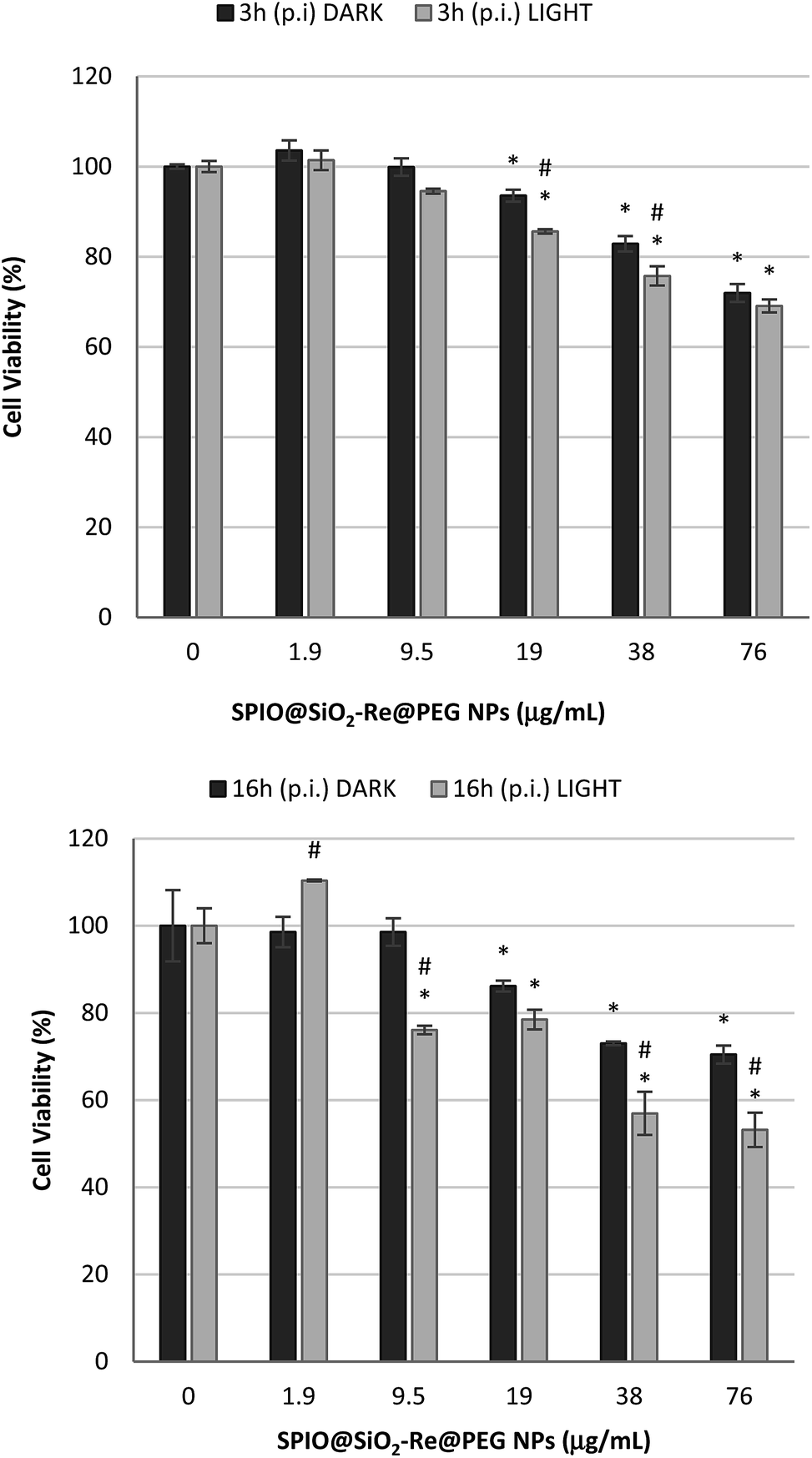 | ||
| Fig. 6 Cell viability (MTT assay) of A549 cells exposed to SPIO@SiO2–Re@PEG NPs for 3 h and 16 h post irradiation (p.i.). Black bars = dark conditions; grey bars = light conditions: cells were exposed to NPs for 4 h, irradiated for 10 min and incubated for 3 h or 16 h again. *Significantly different from control; #significantly different light vs. dark conditions (ANOVA + Fischer LSD test, p < 0.05). | ||
The slight cytotoxic effect observed for cells exposed to the highest NP concentrations in the dark should be attributed to the whole nanostructure, rather than to the sensitizer (i.e. the Re complex). Indeed the [Re(phen)(CO)3(py-upts)]OTf complex alone did not induce any cytotoxicity, as shown by viability tests performed by exposing A549 cells for different times (4 h or 24 h) to increasing concentrations of the complex (see Fig. S11†).
4. Conclusions
A multifunctional nanocomposite, designed for being a bimodal imaging probe that can be exploited also for PDT, has been prepared and characterized. As shown by the results, each component of the nanocomposite preserved the ability to perform its function. The superparamagnetic iron oxide cores, coated by the thick layer of silica and by PEG, to improve stability and stealthiness in physiological conditions, remained able to act as T2 agents for MRI, providing a good contrast ability, as demonstrated by the relaxivity profiles.At the same time, the luminescence properties, as lifetimes and photoluminescence quantum yields, were even improved by the entrapment of the emitting rhenium complex in the silica shell, due to increased rigidity of the environment and to reduced interaction with the water. Finally, the Re complexes anchored to the NPs were still able to act as effective 1O2 photosensitizer.
Preliminary investigation has revealed easy cellular uptake and increased cytotoxicity upon irradiation with respect to the dark, even if with a low efficiency. This might be related to the NP localization at subcellular level, which is known to have strong influence on cell response to photoactivation.21,24,69 Further studies on the internalization mechanism and on the subcellular localization will be necessary to assess the true potential of these nanocomposites. In a parallel way the performances of the diagnostic and therapeutic functionalities integrated in the SPIO@SiO2–Re@PEG NPs can be optimized: the T2 contrast ability would benefit from a larger size of the superparamagnetic core and a smaller thickness of the silica shell, while the luminescence properties as well as the capability of 1O2 generation would be improved by an increased Re loading.
Acknowledgements
The use of instrumentation purchased through the Regione Lombardia – Fondazione Cariplo joint SmartMatLab Project (2013-1766) is gratefully acknowledged. The authors also thank INSTM-Regione Lombardia (project MAGNano) for funding.Notes and references
- See for instance: (a) G. Chen, I. Roy, C. Yang and P. N. Prasad, Chem. Rev., 2016, 116, 2826–2885 CrossRef CAS PubMed; (b) X. Chen, S. S. Gambhir and J. Cheon, Special issue, Theranostic Nanomedicine, Acc. Chem. Res., 2011, 44, 841–1134 Search PubMed.
- E. Duguet, S. Vasseur, S. Mornet and J.-M. Devoisselle, Nanomedicine, 2006, 1, 157–168 CrossRef CAS PubMed.
- A. K. Gupta and A. S. Curtis, J. Mater. Sci.: Mater. Med., 2004, 15, 493–496 CrossRef CAS PubMed.
- K. Ding, L. H. Jing, C. Y. Liu, Y. Hou and M. Y. Gao, Biomaterials, 2014, 35, 1608–1617 CrossRef CAS PubMed.
- G. N. Wang, L. Jin, Y. K. Dong, L. Niu, Y. X. Liu, F. Ren and X. G. Su, New J. Chem., 2014, 38, 700–708 RSC.
- G. N. Wang and X. G. Su, Analyst, 2011, 136, 1783–1798 RSC.
- J. H. Lee, Y. W. Jun, S. I. Yeon, J. S. Shin and J. Cheon, Angew. Chem., Int. Ed., 2006, 45, 8160–8162 CrossRef CAS PubMed.
- Z. Ali, A. Z. Abbasi, F. Zhang, P. Arosio, A. Lascialfari, M. F. Casula, A. Wenk, W. Kreyling, R. Plapper, M. Seidel, R. Niessner, J. Knoll, A. Seubert and W. J. Parak, Anal. Chem., 2011, 83, 2877–2882 CrossRef CAS PubMed.
- Q. Ma, Y. Nakane, Y. Mori, M. Hasegawa, Y. Yoshioka, T. M. Watanabe, K. Gonda, N. Ohuchi and T. Jin, Biomaterials, 2012, 33, 8486–8494 CrossRef CAS PubMed.
- W. Jiang, K. L. Lai, K. X. Liu, R. Xia, F. B. Gao, Y. Wu and Z. W. Gu, Nanoscale, 2014, 6, 1305–1310 RSC.
- (a) M. P. Coogan and V. Fernandez-Moreira, Chem. Commun., 2014, 50, 384–399 RSC; (b) E. Baggaleya, J. A. Weinsteina and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762–1785 CrossRef; (c) K. K.-W. Lo, M.-W. Louie and K. Y. Zhang, Coord. Chem. Rev., 2010, 254, 2603–2622 CrossRef CAS; (d) H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128–6185 RSC; (e) F. L. Thorp-Greenwood, R. G. Balasingham and M. P. Coogan, J. Organomet. Chem., 2012, 714, 12–21 CrossRef CAS; (f) E. Licandro, M. Panigati, M. Salmain and A. Vessières, in Bioorganometallic Chemistry: Applications in Drug Discovery, Biocatalysis, and Imaging, ed. G. Jaouen and M. Salmain, Wiley-VCH, Weinheim, 2015, pp. 341–391 Search PubMed.
- For some pertinent reviews, see: (a) A. Kumar, S.-S. Sun and A. J. Lees, Top. Organomet. Chem., 2010, 29, 1–35 CAS; (b) D. R. Striplin and G. A. Crosby, Coord. Chem. Rev., 2001, 211, 163–175 CrossRef CAS; (c) D. J. Stukfens Jr and A. Vlček, Coord. Chem. Rev., 1998, 177, 127–179 CrossRef; (d) A. J. Lees, Chem. Rev., 1987, 87, 711–743 CrossRef CAS.
- See, for instance: (a) H. C. Bertrand, S. Clède, R. Guillot, F. Lambert and C. Policar, Inorg. Chem., 2014, 53, 6204–6223 CrossRef CAS PubMed; (b) V. Fernández-Moreira, I. Marzo and M. C. Gimeno, Chem. Sci., 2014, 5, 4434–4446 RSC; (c) F. L. Thorp-Greenwood, M. P. Coogan, L. Mishra, N. Kumari, G. Rai and S. Saripella, New J. Chem., 2012, 36, 64–72 RSC; (d) C. Mari, M. Panigati, L. D'Alfonso, I. Zanoni, D. Donghi, L. Sironi, M. Collini, S. Maiorana, C. Baldoli, G. D'Alfonso and E. Licandro, Organometallics, 2012, 31, 5918–5928 CrossRef CAS; (e) G. Gasser, A. Pinto, S. Neumann, A. M. Sosniak, M. Seitz, K. Merz, R. Heumann and N. Metzler-Nolte, Dalton Trans., 2012, 41, 2304–2313 RSC; (f) M.-W. Louie, T. T.-H. Fong and K. K.-W. Lo, Inorg. Chem., 2011, 50, 9465–9471 CrossRef CAS PubMed; (g) K. K. W. Lo, K. Y. Zhang and S. P. Y. Li, Eur. J. Inorg. Chem., 2011, 24, 3551–3568 CrossRef; (h) E. Ferri, D. Donghi, M. Panigati, G. Prencipe, L. D'Alfonso, I. Zanoni, C. Baldoli, S. Maiorana, G. D'Alfonso and E. Licandro, Chem. Commun., 2010, 46, 6255–6257 RSC; (i) M.-W. Louie, T. T.-H. Fong and K. K.-W. Lo, Inorg. Chem., 2011, 50, 9465–9471 CrossRef CAS PubMed; (j) S. James, K. P. Maresca, J. W. Babich, J. F. Valliant, L. Doering and J. Zubieta, Bioconjugate Chem., 2006, 17, 590–596 CrossRef CAS PubMed; (k) K. K.-W. Lo, K. H.-K. Tsang and N. Zhu, Organometallics, 2006, 25, 3220–3227 CrossRef CAS; (l) S. J. Reece, M. R. Seyedsayamdost, J. Stubbe and D. G. Nocera, J. Am. Chem. Soc., 2006, 128, 13654–13655 CrossRef CAS PubMed; (m) K. K.-W. Lo, W.-K. Hui, C.-K. Chung, K. H.-K. Tsang, T. K.-M. Lee, C.-K. Li, J. S.-Y. Lau and D. C.-M. Ng, Coord. Chem. Rev., 2006, 250, 1724–1736 CrossRef CAS.
- (a) M. Mauro, C.-H. Yang, C.-Y. Shin, M. Panigati, C.-H. Chang, G. D'Alfonso and L. De Cola, Adv. Mater., 2012, 24, 2054–2058 CrossRef CAS PubMed; (b) X. Li, D. Zhang, H. Chi, G. Xiao, Y. Dong, S. Wu, Z. Su, Z. Zhang, P. Lei, Z. Hu and W. Li, Appl. Phys. Lett., 2010, 97, 263303–263306 CrossRef; (c) M. Mauro, E. Quartapelle Procopio, Y. Sun, C.-H. Chien, D. Donghi, M. Panigati, P. Mercandelli, P. Mussini, G. D'Alfonso and L. De Cola, Adv. Funct. Mater., 2009, 19, 2607–2614 CrossRef CAS; (d) C. Cebrián, M. Natali, D. Villa, M. Panigati, M. Mauro, G. D'Alfonso and L. De Cola, Nanoscale, 2015, 7, 12000–12009 RSC.
- (a) J. M. Villegas, S. R. Stoyanov, W. Huang and D. P. Rillema, Inorg. Chem., 2005, 44, 2297–2309 CrossRef CAS PubMed; (b) W.-M. Xue, N. Goswami, D. M. Eichhorn, P. L. Orizondo and D. P. Rillema, Inorg. Chem., 2000, 39, 4460–4467 CrossRef CAS.
- (a) L. Sacksteder, A. P. Zipp, E. A. Brown, J. Streich, J. N. Demas and B. A. DeGraff, Inorg. Chem., 1990, 29, 4335–4340 CrossRef CAS; (b) L. Wallace and D. P. Rillema, Inorg. Chem., 1993, 32, 3836–3843 CrossRef CAS.
- (a) M. Panigati, D. Donghi, M. Mauro, P. Mercandelli, P. Mussini, L. De Cola and G. D'Alfonso, Coord. Chem. Rev., 2012, 256, 1621–1643 CrossRef CAS; (b) Y.-H. Tseng, D. Bhattacharya, S.-H. Lin, P. Thanasekaran, J.-Y. Wu, L.-W. Lee, M. Sathiyendiran, M.-L. Ho, M.-W. Chung, K.-C. Hsu, P.-T. Chou and K.-L. Lu, Inorg. Chem., 2010, 49, 6805–6807 CrossRef CAS PubMed; (c) E. Quartapelle Procopio, M. Mauro, M. Panigati, D. Donghi, P. Mercandelli, A. Sironi, G. D'Alfonso and L. De Cola, J. Am. Chem. Soc., 2010, 132, 14397–14399 CrossRef CAS PubMed; (d) D. Donghi, G. D'Alfonso, M. Mauro, M. Panigati, P. Mercandelli, A. Sironi, P. Mussini and L. D'Alfonso, Inorg. Chem., 2008, 47, 4243–4255 CrossRef CAS PubMed.
- S. Carron, M. Bloemen, L. Vander Elst, S. Laurent, T. Verbiest and T. N. Parac-Vogt, J. Mater. Chem. B, 2015, 3, 4370–4376 RSC.
- K. Wähler, A. Ludewig, P. Szabo, K. Harms and E. Meggers, Eur. J. Inorg. Chem., 2014, 807–811 CrossRef PubMed.
- A. Leonidova, V. Pierroz, R. Rubbiani, J. Heier, S. Ferrari and G. Gasser, Dalton Trans., 2014, 43, 4287–4294 RSC.
- A. Kastl, S. Dieckmann, K. Wähler, T. Völker, L. Kastl, A. L. Merkel, A. Vultur, B. Shannan, K. Harms, M. Ocker, W. J. Parak, M. Herlyn and E. Meggers, ChemMedChem, 2013, 8, 924–927 CrossRef CAS PubMed.
- A. A. Abdel-Shafi, J. L. Bourdelande and S. S. Ali, Dalton Trans., 2007, 2510–2516 RSC.
- F. Ragone, H. H. Martinez Saavedra, P. M. David Gara, G. T. Ruiz and E. Wolcan, J. Phys. Chem. A, 2013, 117, 4428–4435 CrossRef CAS PubMed.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- S. Wang, R. Gao, F. Zhou and M. Selke, J. Mater. Chem., 2004, 14, 487–493 RSC.
- R. R. Allison, H. C. Mota and C. H. Shibata, Photodiagn. Photodyn. Ther., 2004, 1, 263–277 CrossRef CAS PubMed.
- D. K. Chatterjee, L. S. Fong and Y. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1627–1637 CrossRef CAS PubMed.
- J. P. Tardivo, A. D. Giglio, C. S. Oliveira, D. S. Gabrielli, H. C. Junqueira, D. B. Tada, D. Severino, R. F. Turchiello and M. S. Baptista, Photodiagn. Photodyn. Ther., 2005, 2, 175–191 CrossRef CAS PubMed.
- K. Ishiyama, K. Nakamura, H. Ikai, T. Kanno, M. Kohno, K. Sasaki and Y. Niwano, PLoS One, 2012, 7, e37871 CAS.
- N. Wang, Z. Zhao, Y. Lv, H. Fan, H. Bai, H. Meng, Y. Long, T. Fu, X. Zhang and W. Tan, Nano Res., 2014, 7, 1291–1301 CrossRef CAS.
- J. C. Stockert, M. Canete, A. Juarranz, A. Villanueva, R. W. Horobin, J. I. Borrell, J. Teixido and S. Nonell, Curr. Med. Chem., 2007, 14, 997–1026 CrossRef CAS PubMed.
- O. Planas, T. Gallavardin and S. Nonell, Chem. Commun., 2015, 51, 5586–5589 RSC.
- Y. You and W. Nam, Chem. Soc. Rev., 2012, 41, 7061–7084 RSC.
- A. Kastl, A. Wilbuer, A. L. Merkel, L. Feng, P. Di Fazio, M. Ocker and E. Meggers, Chem. Commun., 2012, 48, 1863–1865 RSC.
- S. Takizawa, R. Aboshi and S. Murata, Photochem. Photobiol. Sci., 2011, 10, 895–903 CAS.
- C. Zhou and X. Zhao, J. Organomet. Chem., 2011, 696, 3322–3327 CrossRef CAS.
- W. Wu, P. Yang, L. Ma, J. Lalevée and J. Zhao, Eur. J. Inorg. Chem., 2013, 228–231 CrossRef CAS.
- S. S. Lucky, K. C. Soo and Y. Zhang, Chem. Rev., 2015, 115, 1990–2042 CrossRef CAS PubMed.
- Y.-K. Peng, C.-W. Lai, C.-L. Liu, H.-C. Chen, Y.-H. Hsiao, W.-L. Liu, K.-C. Tang, Y. Chi, J.-K. Hsiao, K.-E. Lim, H.-E. Liao, J.-J. Shyue and P.-T. Chou, ACS Nano, 2011, 5, 4177–4187 CrossRef CAS PubMed.
- C.-W. Lai, Y.-H. Wang, C.-H. Lai, M.-J. Yang, C.-Y. Chen, P.-T. Chou, C.-S. Chan, Y. Chi, Y.-C. Chen and J.-K. Hsiao, Small, 2008, 4, 218–224 CrossRef CAS PubMed.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed.
- L. Gao, M. A. Peay and T. G. Gray, Chem. Mater., 2010, 22, 6240–6245 CrossRef CAS.
- (a) N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26–49 CrossRef CAS PubMed; (b) F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharmacol., 2008, 5, 505–515 CrossRef CAS PubMed; (c) L. Sironi, S. Freddi, M. Caccia, P. Pozzi, L. Rossetti, P. Pallavicini, A. Dona, E. Cabrini, M. Gualtieri, I. Rivolta, A. Panariti, L. D'Alfonso, M. Collini and G. Chirico, J. Phys. Chem. C, 2012, 116, 18407–18418 CrossRef CAS; (d) P. Pallavicini, E. Cabrini, G. Cavallaro, G. Chirico, M. Collini, L. D'Alfonso, G. Dacarro, A. Donà, N. Marchesi, C. Milanese, A. Pascale, L. Sironi and A. Taglietti, J. Inorg. Biochem., 2015, 151, 123–131 CrossRef CAS PubMed.
- (a) K. Rahme, L. Chen, R. G. Hobbs, M. A. Morris, C. O'Driscoll and J. D. Holmes, RSC Adv., 2013, 3, 6085–6094 RSC; (b) H. Jans, K. Bonroy, R. De Palma, G. Reekmans, H. Jans, W. Laureyn, M. Smet, G. Borgh and M. Giido, Langmuir, 2008, 24, 3949–3954 CrossRef PubMed.
- (a) S. Schmidt, J. Nitschke and W. Trogler, Inorg. Synth., 1989, 26, 113–117 CrossRef CAS; (b) D. Maggioni, F. Fenili, L. D'Alfonso, D. Donghi, M. Panigati, I. Zanoni, R. Marzi, A. Manfredi, P. Ferruti, G. D'Alfonso and E. Ranucci, Inorg. Chem., 2012, 51, 12776–12788 CrossRef CAS PubMed.
- P. Blondeau, M. Barboiu and E. Petit, React. Funct. Polym., 2015, 86, 259–263 CrossRef CAS.
- S. Benyahya, F. Monnier, M. Taillefer, M. Wong Chi Man, C. Bied and F. Ouazzani, Adv. Synth. Catal., 2008, 350, 2205–2208 CrossRef CAS.
- Rillema et al.15b obtained [Re(L-L)(L′)(CO)3]+ complexes (where L–L is a bpy or a phen bidentate ligand, and L′ is a py like ligand) by a route that involves the tetracarbonyl intermediate [Re(L–L)(CO)4]OTf, formed by stirring the precursor Re(CO)5OTf at room temperature in a non-coordinating solvent, in the presence of the bidentate ligand L–L. The [Re(L–L)(L′)(CO)3]+ complex was subsequently obtained by the addition of one equiv. of the L′ ligand and refluxing for several hours. Another synthetic strategy proposed by Wrighton et al.49 consisted in the substitution of the Cl− ligand with a pyridine in the precursor ReCl(CO)3(L–L), by refluxing in the presence of AgOTf. We obtained the desired product by firstly preparing the precursor complex [Re(phen)(CO)3(OTf)] and then by easily substituting the OTf- anion with the py-upts ligand (Scheme 2).
- S. M. Fredericks, J. C. Luong and M. S. Wrighton, J. Am. Chem. Soc., 1979, 101, 7415–7417 CrossRef CAS.
- J. Park, K. An, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, N.-M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- S. Santra, R. P. Bagwe, D. Dutta, J. T. Stanley, G. A. Walter, W. Tan and R. A. Mericle, Adv. Mater., 2005, 17, 2165–2169 CrossRef CAS.
- H. L. Ding, Y. X. Zhang, S. Wang, J. M. Xu, S. C. Xu and G. H. Li, Chem. Mater., 2012, 24, 4572–4580 CrossRef CAS.
- B. Radi, R. M. Wellard and G. A. George, Soft Matter, 2013, 9, 3262–3271 RSC.
- A. J. Kell, M. L. Barnes, Z. J. Jakubek and B. Simard, J. Phys. Chem. C, 2011, 115, 18412–18421 CAS.
- Also the IR spectra in the ν(CO) region indicate that the Re atom in the [Re(phen)(CO)3(py-upts)]+ complex is more electron rich than in the complex with unsubstituted pyridine (2034, 1934 cm−1, vs. 2037, 1938 cm−1 for the complex with py ligand).
- J. V. Caspar and T. J. Meyer, J. Phys. Chem., 1983, 87, 952–957 CrossRef CAS.
- M. Wrighton and D. L. Morse, J. Am. Chem. Soc., 1974, 96, 998–1003 CrossRef CAS.
- G. Ferrante and S. Sykora, Adv. Inorg. Chem., 2005, 57, 405–470 CrossRef CAS.
- see for instance ref.: (a) V. Amendola, M. Meneghetti, O. M. Bakr, P. Riello, S. Polizzi, S. Fiameni, D. H. Anjum, P. Arosio, T. Orlando, C. de Julian Fernandez, F. Pineider, C. Sangregorio and A. Lascialfari, Nanoscale, 2013, 5, 5611–5619 RSC; (b) W. Di, S. K. P. Velu, A. Lascialfari, C. Liu, N. Pinna, P. Arosio, Y. Sakka and W. Qin, J. Mater. Chem., 2012, 22, 20641–20648 RSC; (c) H. Amiri, R. Bustamante, A. Millán, N. J. O. Silva, R. Piñol, L. Gabilondo, F. Palacio, P. Arosio, M. Corti and A. Lascialfari, Magn. Reson. Med., 2011, 66, 1715–1721 CrossRef CAS PubMed.
- W. Wu, P. Yang, L. Ma, J. Lalevée and J. Zhao, Eur. J. Inorg. Chem., 2013, 228–231 CrossRef CAS.
- D. Maggioni, M. Galli, L. D'Alfonso, D. Inverso, M. V. Dozzi, L. Sironi, M. Iannacone, M. Collini, P. Ferruti, E. Ranucci and G. D'Alfonso, Inorg. Chem., 2015, 54, 544–553 CrossRef CAS PubMed.
- C. Tanielian and C. Wolff, J. Phys. Chem., 1995, 99, 9831–9837 CrossRef CAS.
- S. J. Leach and H. A. Scher, J. Am. Chem. Soc., 1960, 82, 4790–4792 CrossRef CAS.
- W. Tang, H. Xu, R. Kopelman and M. A. Philbert, Photochem. Photobiol., 2005, 81, 242–249 CrossRef CAS PubMed and refs therein.
- The much higher yields reported for some Re complexes20,66 were measured in organic apolar solvents, where non-radiative deactivation pathways of the excited states are less available.
- Z. Yi, J. Zhao, J. Sun, S. Guo and H. Zhang, Dalton Trans., 2013, 42, 2062–2074 RSC.
- D. B. Tada, L. L. R. Vono, E. L. Duarte, R. Itri, P. K. Kiyohara, M. S. Baptista and L. M. Rossi, Langmuir, 2007, 23, 8194–8199 CrossRef CAS PubMed.
- W. Denk, J. Strickler and W. Webb, Science, 1990, 248, 73–76 CAS.
- N. L. Oleinick, R. L. Morris and I. Belichenko, Photochem. Photobiol. Sci., 2002, 1, 1–21 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, DLS and TGA analyses, NMR, IR and UV-vis spectra, confocal microscope images and MTT test graphs are presented. See DOI: 10.1039/c6ra04332a |
| This journal is © The Royal Society of Chemistry 2016 |