
A mesoporous Ni3N/NiO composite with a core–shell structure for room temperature, selective and sensitive NO2 gas sensing†
Mingming Zoua,
Hu Mengb,
Fengdong Qua,
Liang Feng*b and
Minghui Yang*a
aDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, P. R. China. E-mail: myang@dicp.ac.cn
bKey Laboratory of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, P. R. China. E-mail: fengl@dicp.ac.cn
First published on 25th April 2016
Abstract
A highly crystallized mesoporous Ni3N with core–shell structure was prepared from a green and template free method. The Ni3N was characterized by using powder X-ray diffraction (PXRD), scanning electron microscopy (SEM), Brunauer–Emmett–Teller (BET) surface area analysis, and the measured Ni3N has a specific surface area of 85.89 m2 g−1 and a narrow pore size distribution centered at 5 nm. After air oxidization, the Ni3N/NiO composite was further evaluated for NO2 gas sensing at room temperature, and the Ni3N/NiO-based sensor exhibited excellent selectivity for NO2 gas. A good linear relationship between the sensor response and the NO2 gas concentrations was observed. The enhanced NO2 sensing performance can be attributed to the highly crystallized mesoporous and core–shell structures of the synthesized Ni3N/NiO.
Introduction
Sensors with low working temperature, high sensitivity and good selectivity are in great demand not only for environmental monitoring, but also for human health. Owing to sensitivity, rapid response and inherent stability, a variety of nanostructured metal oxide semiconductor based gas sensors have been used to detect flammable, harmful and toxic gases such as NO2, CO, H2S, volatile organic compounds (VOCs).1 NiO is an important native p-type semiconductor has attracted considerable attention due to its unique properties such as the relative wide band gap (Eg = 3.6–4.0 eV), and a measurable electrical conductivity change accompanied with chemical reactions on the surface of the materials. Sensors with various morphologies of NiO (nanoparticles, nanosheets, nanotubes, fibers, hollow spheres, nanowires),2,3 doped-NiO (Pt, Au, Fe, and Zn),4,5 and NiO mixed with other oxide semiconductors (ZnO, SnO2, and PdO)6,7 have been developed in recent years. However, NiO based gas sensor for room temperature applications is still a challenge, showing a great technological and scientific interest.8So far several preparation methods have been reported for synthesizing nickel nitrides, including rapid and exothermic solid-state metathesis (SSM) reactions, supercritical synthesis, laser-aided synthesis, etc.9–12 Among them, SSM and supercritical synthesis do not require temperatures higher than 500 K. On the flip side, such techniques almost inevitably introduce by-products as impurities, and hence such products need further treatment. Typically phase pure metastable nitride (Ni3N) cannot be easily prepared using nitridation of metal powders in NH3 flow, because of their thermal instability. Currently synthesis of Ni3N using a green, cost-effective approach, that does not use high pressure, continues to be a challenge.
In this study, we present a facile, scalable, and controllable synthesis of large-surface-area mesoporous Ni3N with core–shell structure. The Ni3N was characterized by using powder X-ray diffraction (PXRD), scanning electron microscopy (SEM), Brunauer–Emmett–Teller (BET) surface area analysis, and the measured Ni3N has a specific surface area of 85.89 m2 g−1 and a narrow pore size distribution centered at 5 nm. Further, after air oxidation, the as-synthesized Ni3N/NiO composite is found to have excellent gas sensing properties for the room-temperature NO2 detection. As is well known, the semiconductor metal oxide based gas sensors have been widely used to detect a variety of volatile organic compounds (VOCs), however, the high working temperature not only increases the power consumption, but brings risks of explosion. Thus, the room temperature gas sensors are still highly expected, especially for some toxic gases, such as NO2, CO, H2S, etc. Generally, researchers combine graphene oxide and metal oxide to obtain room temperature sensing properties, the complicated synthesis procedure limits its further application. In this work, based on our mesoporous Ni3N material, by simple air oxidation, a Ni3N/NiO composite with large-surface-area was easily achieved, and was utilized as sensing materials. The Ni3N/NiO composite-based sensor exhibited excellent responsive sensitivity and good selectivity to NO2 at room temperature.
Experimental
(a) Materials and synthesis methods
All chemicals were purchased from Aldrich without further purification. Ni(SO4)2 (nickel(II) sulfate hexahydrate, 99%) was used as the nickel sources. The nickel salt and urea were readily mixed in ethanol (50 mL) and water (30 mL) solution. The concentration of nickel salt and urea was fixed at 0.06 mol L−1 and 0.30 mol L−1, respectively. The solvothermal treatment was carried out by transferring the resulting solutions into a Teflon-lined autoclave and heated at 160 °C for 18 h. The resulting precipitate was collected by washing with ethanol and water, centrifugal separation, and dried in air. NiO was obtained by calcining the dried precipitate in air at 500 °C for 2 h. The resulting NiO powder was placed in a quartz boat. This boat was then placed in a quartz tube with airtight, stainless steel end-caps that have welded valves and connections to input and output gas lines. The quartz tube was then placed in a tube furnace, and the appropriate connections to the gas sources were made. An argon gas flow through the tube was used for 15 min to expel the air remaining in the tube before establishing the flow of ammonia gas through the tube. The Ni3N/NiO sample was then heated in the tube to 350 °C at 4 °C min−1. After 2 h elapsed at 350 °C, the furnace power was turned off and the product was cooled to room temperature in 4 h under an ammonia gas flow. Before the quartz tube was taken out of the tube furnace, an argon gas flow through the tube was used to expel the ammonia gas remaining in the tube.(b) Materials characterization
The obtained samples were examined by Rigaku MiniFlex 600 powder X-ray diffractometer. Crystal structures of all the resulting samples were confirmed by PXRD profiles using the GSAS package. The morphology of materials were examined by scanning electron microscopy instrument (JSM-7800F, Japan). Surface area measurements were performed by nitrogen adsorption using Brunauer–Emmet–Teller (BET) area method on Accelerated Surface Area and Porosimetry System (ASAP 2420) to obtain the value of specific surface area, pore volume and mean pore size.(c) Sensor fabrication and gas sensing measurements
The synthesized Ni3N/NiO product was dispersed in absolute anhydrous ethanol at around 5 mg mL−1 by ultrasonication, and then 1 μL of the mixture was dropped onto an Al2O3 ceramic tube (4 mm in length, 1.2 mm in external diameter, and 0.8 mm in internal diameter). After solvent evaporation, a Ni3N/NiO layer formed on the surface of the ceramic tube. A pair of gold electrodes was attached to each end of the ceramic tube. The gas sensing performances of Ni3N/NiO-based sensor were carried out in a homemade sensor testing system (a cylindrical glass chamber with a volume of 100 mL). A gas mixing line equipped with mass flow controllers was designed to prepare the targeted gas at specific concentrations and relative humidity (RH) in the testing chamber, which was shown in Fig. S1 (given in the (ESI†)). The resistance changes of Ni3N/NiO-based sensor in air or targeted gas were monitored by a high-resistance meter (Victor, 86E, China), and the response signal (S) of Ni3N/NiO-based sensor was defined as the Ra/Rg ratio (Ra, resistance in air; Ra, resistance in tested gas). Before any sensing experiment, the sensor testing system was purged with air at 33% RH until a constant resistance was obtained. The sensor was then exposed to targeted gas at same RH and air flow alternately. The time taken by the sensor to achieve 90% of the total resistance change was defined as the response time in the case of response (target gas adsorption) or the recovery time in the case of recovery (target gas desorption).Results and discussion
(a) Synthesis of Ni3N/NiO composite
All the reaction conditions for the synthesis of Ni(HCO3)2 and Ni(OH)2 are summarized in Table S1.† The PXRD patterns were used to identify the crystal structure and composition of these precursor samples. As shown in Fig. S2 (ESI†), the PXRD peak positions for all the samples (samples a–e and g in Table S1†) are matched well with the hexagonal 3Ni(OH)2·2H2O phase (ICDD no. card 22-0444) with the lattice parameters a = 3.08 Å and c = 23.41 Å. Because of the weakly alkaline medium created by stepwise hydrolysis of urea in the solvothermal reactions, β-Ni(OH)2 phase, which is generally synthesized from alkaline pH condition, is not observed in this case. Although the same Ni sources with different synthesis conditions are found to form an identical crystal phase, slight shifts occur in the diffraction peaks, which can be ascribed to the small difference in the α-Ni(OH)2 lattice.13 A broad and weak diffraction peak is found in Fig. S2 (ESI†). This result suggests that particles with small domain size were formed. As shown in Fig. S3 (ESI†), the PXRD peak positions for the two precursor samples (samples f and h in Table S1†) are matched well with the cubic Ni(HCO3)2 phase (ICDD no. card 15-0782) with the lattice parameters a = 8.3830 Å. When the solvothermal reactants was maintained at 160 °C for 18 h, 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 urea:Ni mol ratio with H2O or H2O and alcohol, the formed α-Ni(OH)2 was completely converted into Ni(HCO3)2. It is due to the fact that when concentration of carbonate anions reaches a certain degree at high temperature for a long enough reaction time. Thus, in the progress of solvothermal reaction, carbon dioxide released by urea has not only effect on the growth of Ni(OH)2, but also conducive to the formation of Ni(HCO3)2.
:1 urea:Ni mol ratio with H2O or H2O and alcohol, the formed α-Ni(OH)2 was completely converted into Ni(HCO3)2. It is due to the fact that when concentration of carbonate anions reaches a certain degree at high temperature for a long enough reaction time. Thus, in the progress of solvothermal reaction, carbon dioxide released by urea has not only effect on the growth of Ni(OH)2, but also conducive to the formation of Ni(HCO3)2.
Sample h was used as a precursor due to the hollow and mesoporous feature. Upon calcination of 500 °C for 2 h, Ni(HCO3)2 was converted into NiO. This sample display a monoclinic (a lattice parameter a = 5.1096 Å, b = 2.9560 Å, c = 2.9510 Å) NiO phase (ICDD no. 65-6920) by PXRD patterns without any impurity. The obtained NiO samples were all in black color because of the presence of Ni vacancies, indicating their nonstoichiometric (Ni1−xO) nature.14 After ammonolysis of NiO at 400 °C for 2 h, only clear diffraction peaks for Ni3N were observed by PXRD in Fig. 1. This confirms the phase purity of Ni3N, and also suggests highly crystallized ammonolysis product formed. The Ni3N crystallizes in the space group P6322 with a refined lattice parameter of a = 4.6185(2) Å and c = 4.3022(2) Å. And it shows relatively broad diffraction peaks due to the crystalline domain sizes of 71 nm (calculated by Rietveld fit of the broadened Lorentzian X-ray line shape in GSAS of Fig. S4†). After ammonolysis of NiO at 350 °C for 2 h, an interphase of Ni3N/NiO formed for the incomplete nitride.
 | ||
| Fig. 1 XRD patterns of MHCSS-Ni(HCO3)2 microsphere, the resultant NiO, Ni3N/NiO and Ni3N. | ||
(b) Influences of reaction conditions on the shape of Ni(HCO3)2 or Ni(OH)2
In solvothermal reaction, long reaction time would increase the crystallinity of the products, and generally with increased particle size.15,16 In our case, for the synthesis of Ni(OH)2 and Ni(HCO3)2 nanocrystals, no pronounced influence on particle size was found with reaction time. However, relatively long reaction time gives a rough surface of the products (sample c, d, g and h in Fig. S5†). In contrast, prolonged heating resulted in mass transport via Ostwald ripening (a spontaneous process involving the growth of large precipitates at the expense of smaller precipitates, caused by energetic factors) results in a generation of interior spaces in inorganic nanostructures.Reaction temperature affects both the nucleation and the growth rates in solvothermal synthesis of nanocrystals. The particle sizes of products increased with reaction temperature; going from 100 to 160 °C resulted in increased in particle size from ∼1 μm for sample a, b to ∼1.5–2.5 μm for sample g, h. Generally the higher of the temperatures, the bigger of final particle size is for an identical reaction time. Thus in order to control the crystallinity and particle size of the products, it is necessary to maintain an appropriate temperature in the solvothermal reaction.
The morphology of nanocrystals can be also controlled by varying the urea:Ni mol ratio. As shown in Fig. S5 (a, c, e and g) (ESI†), the morphology of the nanocrystals are all quasi-spherical. Various 3-D Ni(OH)2 microspheres consisting of 1D nanowires with relatively irregular morphology; parts of which are collapsed are obtained. As shown in Fig. S5 (b, d, f, h) (ESI†), the morphology of 3-D Ni(OH)2 or Ni(HCO3)2 microspheres are maintained, but 2D nanoplates or 3D nanocubic agglomerate on the surface. Specially, more cubic-like nanocrystals can be found on the surface in Fig. S5 (h) (ESI†). The reaction solutions are also important for controlling the surface morphology and sphere diameter. For the same reaction using different solvents, different target product with various morphologies and particle size dispersion are observed (sample f, h in Fig. S3 (f and h)).†
(c) Formation mechanisms of the mesoporous and core–shell structures
The mesoporous Ni(HCO3)2 microsphere with heterogeneous core–shell structure (named MHCSS-Ni(HCO3)2 microsphere) (sample h) is obtained by free precipitation from solvothermal reaction at 160 °C for 18 h. At 70 °C, NH3 and excess CO2 released by excessive urea hydrolysis, and the presence of weak alkaline solution creates a mass of nucleation sites for self-assembly of nanocube Ni(HCO3)2 (ref. 17) (Fig. 2a). Here, urea acts not only as a mineralizing agent to induce the formation and crystal growth of α-Ni(HCO3)2, but also as a morphology controller to mediate the self-assembly of α-Ni(HCO3)2 nanocubic structures. With the increase of temperature and reaction time, a secondary growth and agglomeration of nanocube Ni(HCO3)2 takes place, and smaller crystallites would eventually dissolve into solution and regrow on the larger ones (called Ostwald ripening, Fig. 2b1 and b2). Then, the solid evacuation starts at a particular region underneath the immediate surface layer, which divides the α-Ni(HCO3)2 solid microsphere into two discrete regions and leaves hollow spaces (Fig. 2c1 and c2). If the hollowing proceeds in an asymmetrical manner, the resultant semi-hollow core–shell structures are basketlike (Fig. 2d1 and d2), which results in a tumbler-type mass imbalance.18 In this study, MHCSS-Ni3N/NiO microsphere was derived from calcining Ni(HCO3)2 (sample h) in air followed by nitriding in NH3 flow. Under relatively low temperature and short calcination, the final morphologies of Ni3N/NiO (Fig. 3a, c and d) maintained from Ni(HCO3)2 (Fig. S3h†). In addition, Fig. 3b shows the shell of MHCSS-Ni3N/NiO microsphere after ultrasonic irradiation for 1 h which gives an intuitively understanding of the core shell structure of the product. | ||
| Fig. 2 Mechanism of the core shell formation of the MHCSS- Ni(HCO3)2 microsphere. | ||
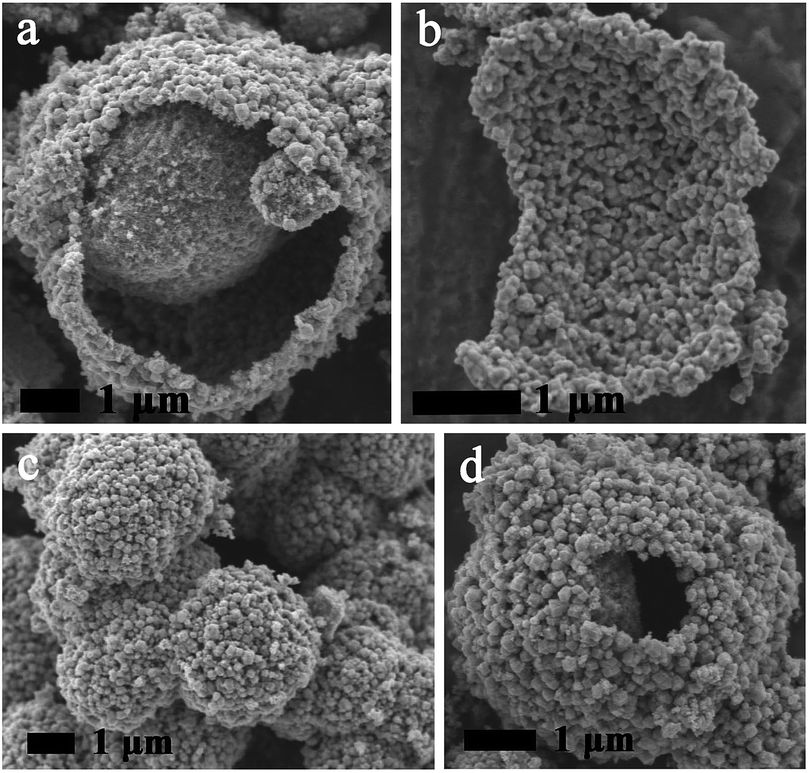 | ||
| Fig. 3 (a–d) SEM of MHCSS- Ni3N/NiO microsphere. | ||
(d) Specific surface area of MHCSS-Ni3N/NiO microsphere
The textural properties of MHCSS-Ni3N/NiO microsphere were analyzed by N2 adsorption. As shown in Fig. 4, type IV isotherms with a sharp capillary condensation step at high relative pressures (P/P0 ≈ 0.7–1.0) and H1 type hysteresis loops are observed for the analyzed sample.19 This indicates a relatively large pore sizes with uniform pore size distribution of the sample. The MHCSS-Ni3N/NiO microsphere had a specific surface area of 85.89 m2 g−1 and a narrow pore size distribution centered at 5 nm. | ||
| Fig. 4 Nitrogen sorption isotherms of the MHCSS- Ni3N/NiO microsphere by BET measurements. | ||
(e) Gas-sensing property of MHCSS-Ni3N/NiO microsphere
The measurable resistance of the as-synthesized Ni3N/NiO indicates its semiconducting property and thus can be tentatively utilized as sensing material. The gas sensor was fabricated according to previous reports.20,21 Fig. 5a shows the dynamic resistance changes of the as-fabricated Ni3N/NiO-based sensor to various concentrations of NO2 under 33% RH at room temperature. The sensor resistance decreases with the exposure of NO2, indicating the p-type response of the sensor. The sensing time of each NO2 concentration was fixed at 10 min for further measurements and comparison. Obviously, as the NO2 concentration goes up, the resistance of the sensor keeps decreasing. A linear fitting curve of the sensor response versus the NO2 concentration in the range of 1 to 3 ppm is obtained, as shown in the insert, and the correlation coefficient R2 of the fitting curve is 0.9954, indicating a good linearity. The detection limit for the Ni3N/NiO composites-based sensor is estimated to be approximately 300 ppb, when the criterion for NO2 detection is set to S > 1.2.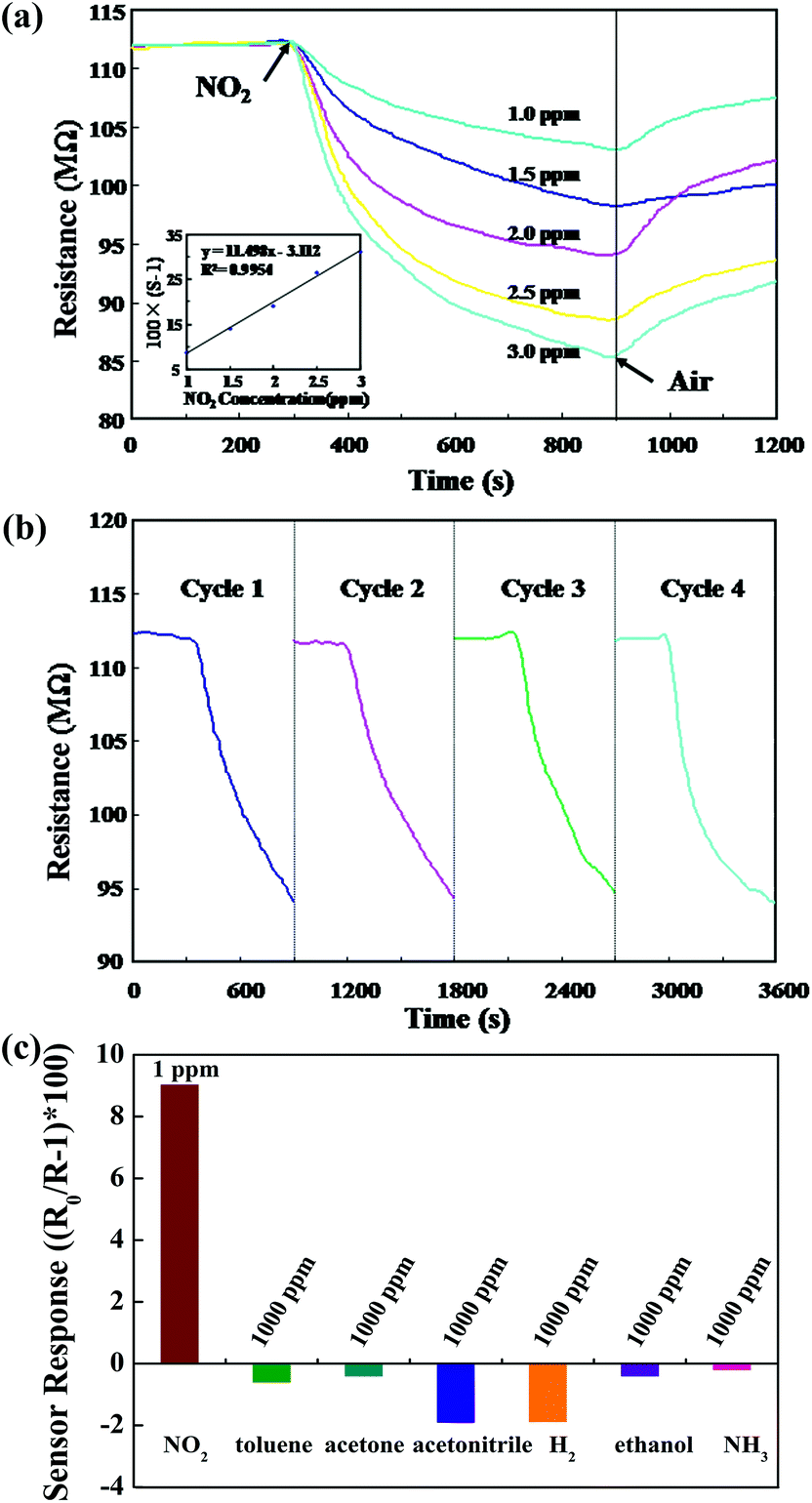 | ||
| Fig. 5 The sensing properties of Ni3N/NiO-based gas sensor for NO2 at room temperature. (a) Kinetic responses of the Ni3N/NiO-based sensor for NO2 sensing at room temperature: insert is the linear fitting curve of the sensor response against NO2 concentration; (b) the evaluation for reproducibility of sensor for 2 ppm NO2; (c) the sensor responses for 1 ppm NO2 and several possible interferential gases at 1000 ppm, including toluene, acetone, acetonitrile, hydrogen, ethanol, and ammonia. | ||
The repeatability of Ni3N/NiO-based sensor was also investigated. The sensor was exposed to 2 ppm NO2 for 10 min under 33% RH at room temperature for four response cycles, as shown in Fig. 5b. All four response cycles present very similar trends after exposure to NO2. The long term stability of the sensor was evaluated by exposure the sensor to 2 ppm of NO2 under 33% RH at room temperature for a period of 2 weeks, and the sensor exhibits good stability and only less than ∼10% variation is observed (Fig. S6, ESI†). Moreover, Ni3N and NiO themselves did not show any sensing properties for NO2 at room temperature. Their mixtures did not present response either (Fig. S7, ESI†). This largely proves that there might be some synergetic effect in our Ni3N/NiO composite, leading the room temperature sensing for NO2.
Selectivity is also an important parameter for gas sensor in practical applications. Fig. 5c reveals the cross-response of the Ni3N/NiO-based sensor to several possible interferential gases, including toluene, acetone, acetonitrile, hydrogen, ethanol, and ammonia. It is clearly observed that the sensor exhibits the largest response to NO2 among these gases, even though the concentration of the other tested gases are thousands times that of NO2. Such results indicate that the sensor exhibits an excellent selectivity toward NO2 under 33% RH at room temperature. The Ni3N/NiO-based sensor exhibits higher response towards NO2 among various oxidizing and reducing gases. This might be due to the high reactivity and large electron affinity (2.28 eV) of NO2, in comparison with pre-adsorbed oxygen (0.43 eV) and other test gases.22 The oxygen deficient sensor surface increases the number of sites for the adsorption of the NO2, thereby making films selective towards NO2 than other gases.
A comparison between the sensing performances of the sensor and literature reports is summarized in Table 1. So far, many literature have reported the gas sensing properties to NO2. It is noteworthy that the sensor fabricated in our work exhibits better sensing performances compared with those reported in the literatures.
| Sensing materials | NO2 (ppm) | Temperature (°C) | Response | Response time (s) | Recovery time (s) | Reference |
|---|---|---|---|---|---|---|
| Ni3N/NiO | 1 | RT | Ra/Rg = 1.09 (cal: (Ra − Rg)/Ra = 8.44%) | 900 | 1200 | This work |
| Al-decorated graphene | 1.2 | 150 | (Ra − Rg)/Ra = 2.89% | About 300 | >900 | Ref. 27 |
| Flexible graphene | 5 | RT | (Ra − Rg)/Ra = 3% | 24 | 50 | Ref. 28 |
| Ozone-treated graphene | 5 | RT | (Ra − Rg)/Ra = 3.7% | About 1000 | About 1800 | Ref. 29 |
| In2TiO3-rutile | 100 | RT | (Ra − Rg)/Rg = 4.04% (cal: (Ra − Rg)/Ra = 3.88%) | 1.33 | 8.78 | Ref. 30 |
| Felexible RGO | 5 | RT | (Ra − Rg)/Ra = 12% | 420 | 1680 | Ref. 31 |
(f) Influences of MHCSS-Ni3N/NiO microsphere on gas-sensing properties
The room-temperature NO2 gas sensing mechanism of Ni3N/NiO belongs to a surface-controlled type, in which the adsorption of atmospheric and NO2 molecules on Ni3N/NiO surface plays an important role.23 In ambient air, the oxygen molecules are adsorbed on Ni3N/NiO surface and trap free electrons from conduction band to generate chemisorbed oxygen species, resulting in an increase of the charge carrier density (holes is the majority charge carriers) and the formation of a conduction layer on the surface of the sensing materials.24 Besides, under room temperature, O2− is the dominant adsorbed oxygen species.25 When exposed to NO2, the high electrophilic NO2 molecules not only capture the electrons from the conductance band (eqn (1)), but also react with the absorbed oxygen species (eqn (2)),26 thus resulting in an further increase of the charge carrier density, which eventually decrease the sensor resistance.23| NO2(g) + e− → NO−2(ads) | (1) |
| NO2(g) + O−2(ads) + 2e− → NO−2(ads) + 2O−(ads) | (2) |
There are two important aspects involved in the response of gas sensors, including recognition by the reactive surface and high electron mobility.32 In this case, the hollow and porous morphologies of the Ni3N/NiO composite could enhance gas sensing performance due to both inner and outer surfaces participating in sensor response; enhancing the number of active sites available for gas adsorption. Defective Ni3N occurring over NiO, post-ammonolysis seems responsible for the observed behavior. NiO is a semiconductor, while Ni3N is metallic with a resistivity of 10.9 × 10−5 Ω m at 300 K, and could overcome the poor electrical conductivity of NiO at room temperature, therefore, the effective electronic interaction between Ni3N and NiO facilitates the gas molecule detection via the resistance change of the hybrid composites, which could further improved the gas-sensing properties of the composite materials.
Apparently, the more massive the activity sites are, the more sensitive the sensor is. Exposure of the mass activity sites can be mainly attributed to the uniform pore size distribution and efficient diffusion through the microspheres during the reaction. Even synergistic effects between shell and cores with rough and curved surfaces that are expected to further extend the activity of the nanostructure. And the large contact area leads to a significant increase in the reactivity, due to the high surface-to-volume ratio. In addition, the hollow space given by the core–shell structure, jointly with its porosity, broaden the possibilities to tune the reactivity, prevents internal active nanoparticles from aggregating, thus leading a good surface accessibility. This means that the amount of oxygen that can be absorbed and ionized is increased. Meanwhile, the diffusion of the test gas can be facilitated and the kinetics of the reaction of the test gas with surface-adsorbed oxygen species could be improved. Second, Ni3N exhibits excellent catalytic activity and acts as an efficient electrocatalyst for oxygen reduction reaction, leading to a significantly higher response.33,34
Conclusions
We have shown that a nickel salt–urea–ethanol and H2O ternary solvothermal system can be used for large-scale synthesis of Ni(HCO3)2 or α-Ni(OH)2. By varying the reaction conditions of temperature, duration and urea to Ni molar ratio, Ni(HCO3)2 or α-Ni(OH)2 microspheres with different morphologies (including 1D nanowires, 2D nanoplates or 3D nanocubic on the surface) have been selectively achieved. Nanostructured NiO has been made upon calcinations of Ni(HCO3)2 or α-Ni(OH)2 microsphere precursors in air at 500 °C for 2 h. MHCSS-Ni3N/NiO microsphere was finally synthesized by ammonolysis of the NiO at 350 °C for 2 h. The Ni3N/NiO-based sensor exhibited an excellent selectivity and a good linear relationship between the sensor response and the NO2 gas concentrations. The enhanced NO2 sensing performance can be attributed to the morphology of the Ni3N/NiO, and the improved electron mobility in the sensing process.Acknowledgements
This work is supported by National Natural Science Foundation of China through grant 21471147, the Instrument Developing Project of CAS and Liaoning Provincial Natural Science Foundation through grant 2014020087. M. Yang would like to thank the National "Thousand Youth Talents" program of China.Notes and references
- T. Wagner, S. Haffer, C. Weinberger, D. Klaus and M. Tiemann, Chem. Soc. Rev., 2013, 42, 4036–4053 RSC.
- E. H. El-Ads, A. Galal and N. F. Atta, J. Electroanal. Chem., 2015, 749, 42–52 CrossRef CAS.
- B. Q. Yuan, C. Y. Xu, L. Liu, Q. Q. Zhang, S. Q. Ji, L. P. Pi, D. J. Zhang and Q. S. Huo, Electrochim. Acta, 2013, 104, 78–83 CrossRef CAS.
- K. E. Toghill and R. G. Compton, Electroanalysis, 2010, 22, 1947–1956 CrossRef CAS.
- L. Q. Rong, C. Yang, Q. Y. Qian and X. H. Xia, Talanta, 2007, 72, 819–824 CrossRef CAS PubMed.
- Y. Ding, Y. Wang, B. K. Li and Y. Lei, Biosens. Bioelectron., 2010, 25, 2009–2015 CrossRef CAS PubMed.
- N. J. Yang, W. Smirnov and C. E. Nebel, Electrochem. Commun., 2013, 27, 89–91 CrossRef CAS.
- Q. N. Abdullah, F. K. Yam, Z. Hassan and M. Bououdina, Sens. Actuators, B, 2014, 204, 497–506 CrossRef CAS.
- P. Kroll, J. Solid State Chem., 2003, 176, 530–537 CrossRef CAS.
- C. I. Ritchie, C. Streb, J. Thiel, S. G. Mitchell, H. N. Miras, D. L. Long, T. Boyd, R. D. Peacock, T. McGlone and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 6881–6884 CrossRef CAS PubMed.
- S. Desmoulins-Krawiec, C. Aymonier, A. Loppinet-Serani, F. Weill, S. Gorsse, J. Etourneau and F. Cansell, J. Mater. Chem., 2004, 14, 228–232 RSC.
- A. Leineweber, H. Jacobs, W. Kockelmann, S. Hull and D. Hinz-Hubner, J. Alloys Compd., 2004, 384, 1–5 CrossRef CAS.
- L. P. Xu, Y. S. Ding, C. H. Chen, L. L. Zhao, C. Rimkus, R. Joesten and S. L. Suib, Chem. Mater., 2008, 20, 308–316 CrossRef CAS.
- F. Fievet, P. Germi, F. D. Bergevin and M. Figlarz, J. Appl. Crystallogr., 1979, 12, 387–394 CrossRef CAS.
- V. F. Puntes, D. Zanchet, C. K. Erdonmez and A. P. Alivisatos, J. Am. Chem. Soc., 2002, 124, 12874–12880 CrossRef CAS PubMed.
- J. I. Park, N. J. Kang, Y. W. Jun, S. J. Oh, H. C. Ri and J. Cheon, ChemPhysChem, 2002, 3, 543–547 CrossRef CAS PubMed.
- J. H. Pan, Q. Z. Huang, Z. Y. Koh, D. Neo, X. Z. Wang and Q. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 6292–6299 CAS.
- B. Liu and H. C. Zeng, Small, 2005, 1, 566–571 CrossRef CAS PubMed.
- S. F. Xie, B. J. Zheng, Q. Kuang, X. Wang, Z. X. Xie and L. S. Zheng, CrystEngComm, 2012, 14, 7715–7720 RSC.
- W. Yang, P. Wan, X. D. Zhou, J. M. Hu, Y. F. Guan and L. Feng, ACS Appl. Mater. Interfaces, 2014, 6, 21093–21100 CAS.
- H. Meng, W. Yang, K. Ding, L. Feng and Y. F. Guan, J. Mater. Chem. A, 2015, 3, 1174–1181 CAS.
- V. Ganbavle, S. Inamdar, G. Agawane, J. Kim and K. Rajpure, Chem. Eng. J., 2016, 286, 36–47 CrossRef CAS.
- L. Huang, Z. P. Wang, J. K. Zhang, J. L. Pu, Y. J. Lin, S. H. Xu, L. Shen, Q. Chen and W. Z. Shi, ACS Appl. Mater. Interfaces, 2014, 6, 7426–7433 CAS.
- A. Tricoli, M. Righettoni and A. Teleki, Angew. Chem., Int. Ed., 2010, 49, 7632–7659 CrossRef CAS PubMed.
- F. H. Saboor, T. Ueda, K. Kamada, T. Hyodo, Y. Mortazavi, A. A. Khodadadi and Y. Shimizu, Sens. Actuators, B, 2016, 223, 429–439 CrossRef CAS.
- X. M. Xu, P. L. Zhao, D. W. Wang, P. Sun, L. You, Y. F. Sun, X. S. Liang, F. M. Liu, H. Chen and G. Y. Lu, Sens. Actuators, B, 2013, 176, 405–412 CrossRef CAS.
- B. Cho, J. Yoon, M. G. Hahm, D.-H. Kim, A. R. Kim, Y. H. Kahng, S.-W. Park, Y.-J. Lee, S.-G. Park and J.-D. Kwon, J. Mater. Chem. C, 2014, 2, 5280–5285 RSC.
- M. W. Jung, S. Myung, W. Song, M.-A. Kang, S. H. Kim, C.-S. Yang, S. S. Lee, J. Lim, C.-Y. Park and J.-O. Lee, ACS Appl. Mater. Interfaces, 2014, 6, 13319–13323 CAS.
- M. G. Chung, D. H. Kim, H. M. Lee, T. Kim, J. H. Choi, D. kyun Seo, J.-B. Yoo, S.-H. Hong, T. J. Kang and Y. H. Kim, Sens. Actuators, B, 2012, 166, 172–176 CrossRef.
- H. Wu, L. Wang, J. Zhou, J. Gao, G. Zhang, S. Xu, Y. Xie, L. Li and K. Shi, J. Colloid Interface Sci., 2016, 466, 72–79 CrossRef CAS PubMed.
- P.-G. Su and H.-C. Shieh, Sens. Actuators, B, 2014, 190, 865–872 CrossRef CAS.
- N. Barsan, D. Koziej and U. Weimar, Sens. Actuators, B, 2007, 121, 18–35 CrossRef CAS.
- K. Xu, P. Chen, X. Li, Y. Tong, H. Ding, X. Wu, W. Chu, Z. Peng, C. Wu and Y. Xie, J. Am. Chem. Soc., 2015, 137, 4119–4125 CrossRef CAS PubMed.
- G. Clavel, V. Molinari, A. Kraupner and C. Giordano, Chem.–Eur. J., 2014, 20, 9018–9023 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra04217a |
| This journal is © The Royal Society of Chemistry 2016 |