
LED-cured self-replenishing hydrophobic coatings based on interpenetrating polymer networks (IPNs)†
Abstract
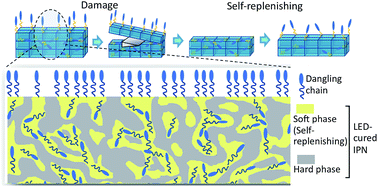
LED-cured IPN-based coatings bearing hydrophobic functional groups have been developed in order to obtain hydrophobic self-replenishing surfaces with improved mechanical properties. Acrylate/epoxide combinations have been chosen to achieve two different Tgs: a lower one to provide sufficient mobility for self-replenishing behavior and a higher one for sufficient hardness. Detailed characterizations of the mechanical and morphological properties of the IPN coatings, in the absence and presence of covalently bonded fluorinated dangling chains, have been performed. Finally, the self-replenishing behavior of these networks with intrinsic hardness has been investigated. This new approach, which is based on IPN and LED technologies could offer self-replenishing functionality for industrial applications, namely in the automotive or aerospace industry.
 Please wait while we load your content...
Please wait while we load your content...

