
Ultrafast pyro-synthesis of NiFe2O4 nanoparticles within a full carbon network as a high-rate and cycle-stable anode material for lithium ion batteries
Preetham P,
Subhalaxmi Mohapatra,
Shantikumar V. Nair,
Dhamodaran Santhanagopalan and
Alok Kumar Rai*
Amrita Centre for Nanosciences and Molecular Medicine, Amrita Vishwa Vidyapeetham, Amrita University, Kochi 682041, Kerala, India. E-mail: alokkumarrai1@gmail.com; alokkumar21223@aims.amrita.edu; Fax: +91-4842 802020; Tel: +91-4842 858750
First published on 5th April 2016
Abstract
NiFe2O4 nanoparticles fully anchored within a carbon network were prepared via a facile pyro-synthesis method without using any conventional carbon sources. The surface morphology was investigated using field-emission scanning electron microscopy, which confirmed the full anchoring of NiFe2O4 nanoparticles within a carbon network. The primary particle size of NiFe2O4 is in the range of 50–100 nm. The influence of the carbon network on the electrochemical performance of the NiFe2O4/C nanocomposite was investigated. The electrochemical results showed that the NiFe2O4/C anode delivered a reversible capacity of 381.8 mA h g−1 after 100 cycles at a constant current rate of 1.0C, and when the current rate is increased to a high current rate of 5.0C, a reversible capacity of 263.7 mA h g−1 is retained. The obtained charge capacity at high current rates is better than the reported values for NiFe2O4 nanoparticles. The enhanced electrochemical performance can be mainly ascribed to the high electrical conductivity of the electrode, the short diffusion path for Li+ ion transportation in the active material and synergistic effects between the NiFe2O4 nanoparticles and carbon network, which buffers the volume changes and prevents aggregation of NiFe2O4 nanoparticles during cycling.
1. Introduction
The nanostructured transition metal oxides which incorporate Li+ ions through a conversion reaction and exhibit large reversible capacities compared to insertion-type electrodes such as graphite or Li4Ti5O12, are considered to be efficient anode materials for high power lithium ion batteries.1,2 Among several transition metal oxides, iron based spinel oxides, such as Fe3O4, as anode materials are significantly researched due to their high theoretical capacity, rich resources, and environmental benignity.3 Spinel type metal oxides have received much attention due to their stable structure and high theoretical capacities. More importantly, partial doping of transition metals into binary iron oxides and modification to ternary ferrites (AFe2O4) also showed a promising and feasible approach to obtain an outstanding electrochemical performance of iron based oxides.4–7 It is believed that these ternary mixed metal oxides could offer better electrochemical performance due to the enhancement in stability of the active phases, the existence of multiple valences of the cations, good electrical properties and the synergetic effect of all these characteristics.8 More importantly, it is also expected that the volume expansion/contraction during charge/discharge in both phases will happen in sequence and reduce the strain of the material. Among various ternary ferrites, NiFe2O4 is quite attractive because both nickel and iron are abundant elements on Earth, are relatively nontoxic and inexpensive and have high thermal stabilities. NiFe2O4 can store 8 moles of Li through a conversion reaction, which corresponds to a theoretical capacity of 915 mA h g−1 (NiFe2O4 + 8Li+ + 8e− ↔ Ni + 2Fe + 4Li2O).9 However, it has been reported that NiFe2O4 exhibits drastic capacity fading and poor cycling stability due to the limited diffusion rate of Li+ ions in the material, poor electrical conductivity and the large volume changes during the Li+ ion insertion/deinsertion processes, leading to pulverization of the electrode and break-down of the electrical contact pathways between adjacent particles.7,9,10 Hence, in order to avoid the above problems, a new strategy should be developed for NiFe2O4 to achieve both a high reversible capacity and long cycling life. It is well-known that nanostructured electrodes offer a high surface area; a large contact area between electrode and electrolyte, which enhances the diffusion kinetics by reducing the diffusion pathway for electronic and ionic transport and ensures a relatively greater number of guest-ions during insertion/deinsertion. Thus, a few attempts such as different synthesis methods to control the morphology and particle sizes have been reported.9–12 In addition, the partial substitution of host-structures with different metal cations to enhance the long-term Li cycling of NiFe2O4 has also been achieved.13 Recently, it is also believed that the fabrication of nanocomposites with graphene nanosheets or carbon materials could be an effective approach to buffer the volume changes due to graphene’s unique properties such as high flexibility, excellent electrical conductivities, large surface area and high mechanical strength.14–17 However, the reaction between oxygen containing functional groups (such as OH, COOH etc.) on the graphene nanosheets and Li+ ions during the insertion/deinsertion processes was believed to be the primary reason for the loss of irreversible capacity.14 On the other hand, a carbon coating is also preferential to improve the interparticle connectivity by developing a conducting network throughout the electrode and suppress excessive particle growth, which can result in improvement of the cycle life.8 However, it is difficult to achieve a uniform dispersion of nanoparticles on a carbon matrix. Hence, a facile synthesis of NiFe2O4 nanoparticles incorporated within a full carbon network in a one step process would be an excellent strategy to improve the electrochemical performance of the NiFe2O4 electrode.Therefore, in the present work, for the first time we have adopted a polyol-assisted pyro-synthesis method to synthesize NiFe2O4 nanoparticles within a full carbon network without using any conventional carbon sources. The polyol solvent acts as a primary fuel and instantly provides ultrahigh exothermic energy to the surroundings during combustion, which leads to thermochemical decomposition of the precursors (endothermic) and nucleation under an oxygen-limited atmosphere by the useful consumption of exothermic energy.18 The feasibility of forming a carbon network under very short reaction times can suppress the particles’ growth and improve the interparticle connectivity. This method has not been investigated with NiFe2O4 nanoparticles to see whether such nanoparticles could be efficiently distributed within a full carbon network, and what would be the electrochemical performance of such a unique nanostructured electrode. The objective is to investigate whether such an electrode would give rise to both high capacity and high stability. The obtained architecture of NiFe2O4 nanoparticles within a full carbon network exhibits a high reversible capacity (768.0 mA h g−1 at 1.0C), a better cycle life (381.8 mA h g−1 at 1.0C after 100 cycles) and an improved rate capability (263.7 mA h g−1 at 5.0C).
2. Experimental
2.1. Materials synthesis
NiFe2O4 nanoparticles within a full carbon network were synthesized by a polyol-assisted pyro-synthesis method and the preparation procedure can be seen in Scheme 1. In a typical synthesis, stoichiometric amounts of nickel(II) acetate tetrahydrate [Ni(OCOCH3)2·4H2O, 98% Sigma Aldrich] and iron(II) acetate [Fe(OCOCH3)2, 95% Sigma Aldrich] were dissolved in 80 ml of diethylene glycol [C4H10O3, 99% Spectrochem] by stirring for 24 h at room temperature. The use of diethylene glycol here acts not only as a solvent and reducing agent but also a carbon source. Subsequently, 50 ml of liquid thinner (Sheenlac, India, benzene free D13X NC thinner) was added to the homogenous solution which was again stirred for 30 min. Then, the resultant solution was transferred onto a hot-plate preheated to 250 °C. The flammable solution was ignited with an electric torch to induce a self-extinguishable combustion process. A black colored powder was directly obtained after the combustion process. Finally, to crystallize the as-prepared powder, it was annealed at 500 °C for 5 h under an argon atmosphere. | ||
| Scheme 1 Schematic illustration of the preparation of NiFe2O4 nanoparticles within a carbon network. | ||
2.2. Materials characterization
The crystal structure of the annealed sample was studied using X-ray diffraction on a Shimadzu X-ray diffractometer with Cu Kα radiation (λ = 1.5406 Å). The particle size and surface morphology of the annealed sample was further studied using field-emission scanning electron microscopy (FE-SEM; S-4800 Hitachi). Field-emission transmission electron microscope (FE-TEM) images were also recorded using a FEI 20 FEG electron microscopy instrument at 200 kV. The samples were first soaked in ethanol and dispersed by ultrasonic vibration before drop casting onto copper grids for FE-TEM examination. The carbon content in the annealed sample was determined by CHN elemental analysis on an Elementar Vario EL III. In order to investigate the nature of the carbon, Raman spectroscopy (Witec Alpha 300 RA) was used with a diode laser as the excitation source.2.3. Electrochemical testing
The electrochemistry of the electrode materials was studied using CR2032 coin type cells. To measure the electrochemical performance, the electrodes were constructed by mixing the annealed NiFe2O4 sample, carbon black and a polyvinylidene fluoride binder in a weight ratio of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:10, respectively in N-methyl-2-pyrrolidone to form a homogenous slurry. Then it was cast on a Cu foil current collector through doctor blading and dried at 80 °C for 12 h in a vacuum oven. Subsequently the slurry was pressed between stainless steel twin rollers to improve the adhesion between the Cu foil and active materials. Lithium foil was used as the counter electrode. 1 M LiPF6 dissolved in an mixture of ethylene carbonate and dimethyl carbonate (molar ratio 1:1, in volume) was used as the non-aqueous electrolyte. A glass fiber was also used as a separator. Cyclic voltammetry was carried out using an Autolab potentiostat (PGSTAT 302 N) with a scan rate of 0.1 mV s−1 within the range of 0–3.0 V. The discharge/charge measurements were performed at room temperature using an Arbin battery tester (BT 2000) in the voltage range of 0.005 V to 3.0 V.
:20:10, respectively in N-methyl-2-pyrrolidone to form a homogenous slurry. Then it was cast on a Cu foil current collector through doctor blading and dried at 80 °C for 12 h in a vacuum oven. Subsequently the slurry was pressed between stainless steel twin rollers to improve the adhesion between the Cu foil and active materials. Lithium foil was used as the counter electrode. 1 M LiPF6 dissolved in an mixture of ethylene carbonate and dimethyl carbonate (molar ratio 1:1, in volume) was used as the non-aqueous electrolyte. A glass fiber was also used as a separator. Cyclic voltammetry was carried out using an Autolab potentiostat (PGSTAT 302 N) with a scan rate of 0.1 mV s−1 within the range of 0–3.0 V. The discharge/charge measurements were performed at room temperature using an Arbin battery tester (BT 2000) in the voltage range of 0.005 V to 3.0 V.
3. Results and discussion
3.1. Structural and morphological analysis
Fig. 1 shows the X-ray diffraction pattern of the annealed NiFe2O4 sample. The XRD pattern displays the majority of the peaks of cubic spinel NiFe2O4 (JCPDS card no. 010742081) with a small fraction of crystalline Fe2O3 (JCPDS card no. 330664) and Ni (JCPDS card no. 011260). It is highly probable that the as-prepared NiFe2O4 sample was slightly reduced to Fe2O3 and Ni metallic phases during annealing under a reduced atmosphere. As per the stoichiometry, 1 mol nickel needs 2 mol iron to produce NiFe2O4. It is believed that a small fraction of NiO is reduced to Ni during annealing and consequently Fe2O3 did not have access to a full amount of NiO to form the stoichiometric NiFe2O4, which leads to the segregation of Fe2O3 as an extra phase within the sample.19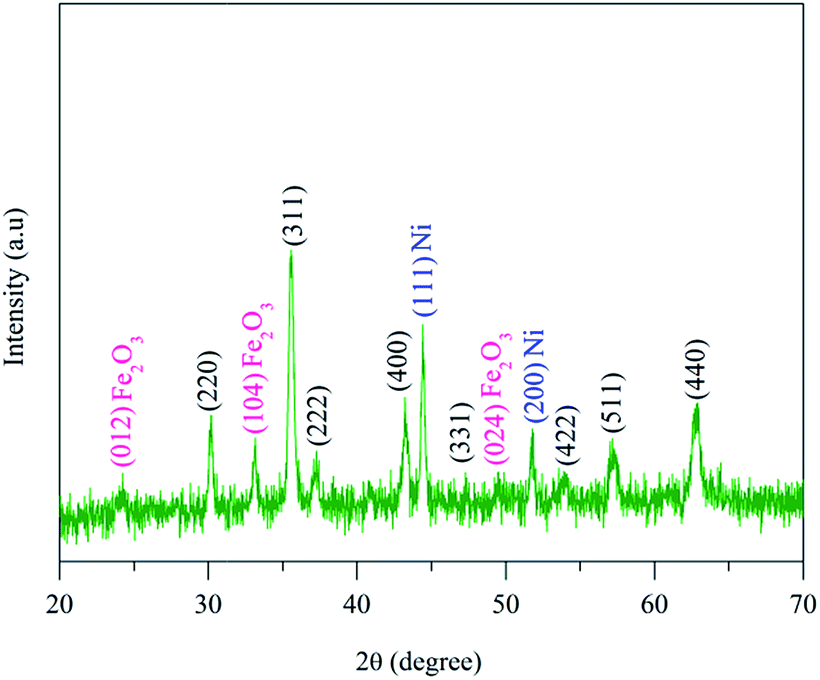 | ||
| Fig. 1 X-ray powder diffraction pattern of annealed NiFe2O4 nanoparticles. | ||
Fig. 2(a) represents a typical FE-SEM image of the annealed NiFe2O4/C nanocomposite. From the image, it is obvious that the NiFe2O4 nanoparticles are highly agglomerated. Furthermore, these NiFe2O4 agglomerates appear to be embedded in a carbon matrix, which can be explained by the interconnection of the NiFe2O4 nanoparticles by carbon. The primary particle size is ranging from 50–100 nm. In order to clearly know the decoration of carbon in the sample and the distribution of nanoparticles within the carbon network, FE-TEM analysis was further investigated. Fig. 2(b)–(d) shows the FE-TEM images of the annealed NiFe2O4/C sample obtained at different magnifications. The FE-TEM images clearly show two distinct regions in the sample. The color contrast between the light gray edge and the dark centre is easily observed. The region with the dark centre is the NiFe2O4 nanoparticles, whereas the light gray region is the carbon network in the sample.18 The low magnification FE-TEM images in Fig. 2(b) and (c) show that the NiFe2O4 nanoparticles are almost uniformly decorated within the carbon network without severe aggregation. More importantly, the inset images of Fig. 2(b) and (c) clearly reveal that no freestanding NiFe2O4 nanoparticles are found in this sample. Furthermore, the high magnification FE-TEM image in Fig. 2(d) markedly exhibits the carbon network between the nanoparticles and the small sized NiFe2O4 nanoparticles are closely anchored within the carbon network. The small sizes and uniformity of these nanoparticles are maintained even after the sonication process during the preparation of the TEM specimen, which apparently shows the strong interaction between the nanoparticles and the carbon sheet. The underlying conductive carbon network would benefit fast electron transfer and maintain structural integrity during the volume change.
 | ||
| Fig. 2 (a) FE-SEM and (b–d) FE-TEM images of NiFe2O4/C nanocomposite. | ||
CHN analysis is performed to determine the accurate percentage of carbon in the annealed sample. CHN analysis of the annealed NiFe2O4/C nanocomposite indicates that the amount of carbon is 4.7%, which would account for a carbon network in the sample, as observed in FE-TEM images of Fig. 2.
Raman spectroscopy is further used to determine the degree of graphitization of the carbon in the annealed sample of NiFe2O4/C. Fig. 3 shows a Raman spectrum of the annealed NiFe2O4/C nanocomposite between the wavenumbers of 500 and 2500 cm−1. As shown in Fig. 3, two bands at wavenumbers of 1358.7 cm−1 and 1596.2 cm−1 are observed, which can be assigned to the D-band and G-band of carbon, respectively. Generally, the D-band is associated with disordered sp3 carbon and defects such as topological defects, dangling bonds, and vacancies, whereas the G-band corresponds to ordered sp2 hybridized carbon.5 The ratio between the D and G band (ID/IG) is used to evaluate the disordered and ordered crystal structures of carbon. Here, the ID/IG ratio for the NiFe2O4/C sample is 0.85, which clearly indicates the good graphitization behaviour of the carbon in the sample.20,21
 | ||
| Fig. 3 Raman spectrum of the NiFe2O4/C nanocomposite. | ||
3.2. Electrochemical performance
Fig. 4(a) shows the cyclic voltammograms of the NiFe2O4/C electrode at a scan rate of 0.1 mV s−1 for the first six cycles in the voltage range of 0–3.0 V (vs. Li/Li+). The first cycle is substantially different from the second one. In the first cathodic scan, a weak cathodic peak at 1.48 V is observed, which could be attributed to the insertion of Li+ ions into the NiFe2O4 electrode,10 whereas the broad intense peak at 0.57 V corresponds to the initial reduction of Ni2+ and Fe3+ to metallic Ni0 and Fe0 nanoparticles along with the formation of a solid electrolyte interface (SEI) layer and amorphous Li2O.10,11,17 However, in the subsequent cycles, the cathodic peak shifts to a higher potential and clearly separates into two peaks at 0.98 V and 0.84 V, indicative of different electrochemical reactions governing the two processes. The large decrease in the peak intensity and integrated area between the first cycle and the following cycles is consistent with a low initial coulombic efficiency, indicating a capacity loss due to the electrolyte decomposition and SEI film formation. Meanwhile, a broad anodic peak around ∼1.64 V could be attributed to the oxidation of metallic Ni0 and Fe0 into Ni2+ and Fe3+, and this peak slightly shifts positively to 1.68 V in the subsequent cycles.10,11,17 Remarkably, all the characteristic peaks and their integrated areas and shapes remained the same after the first cycle, suggesting reversible reduction and oxidation of the NiFe2O4/C electrode.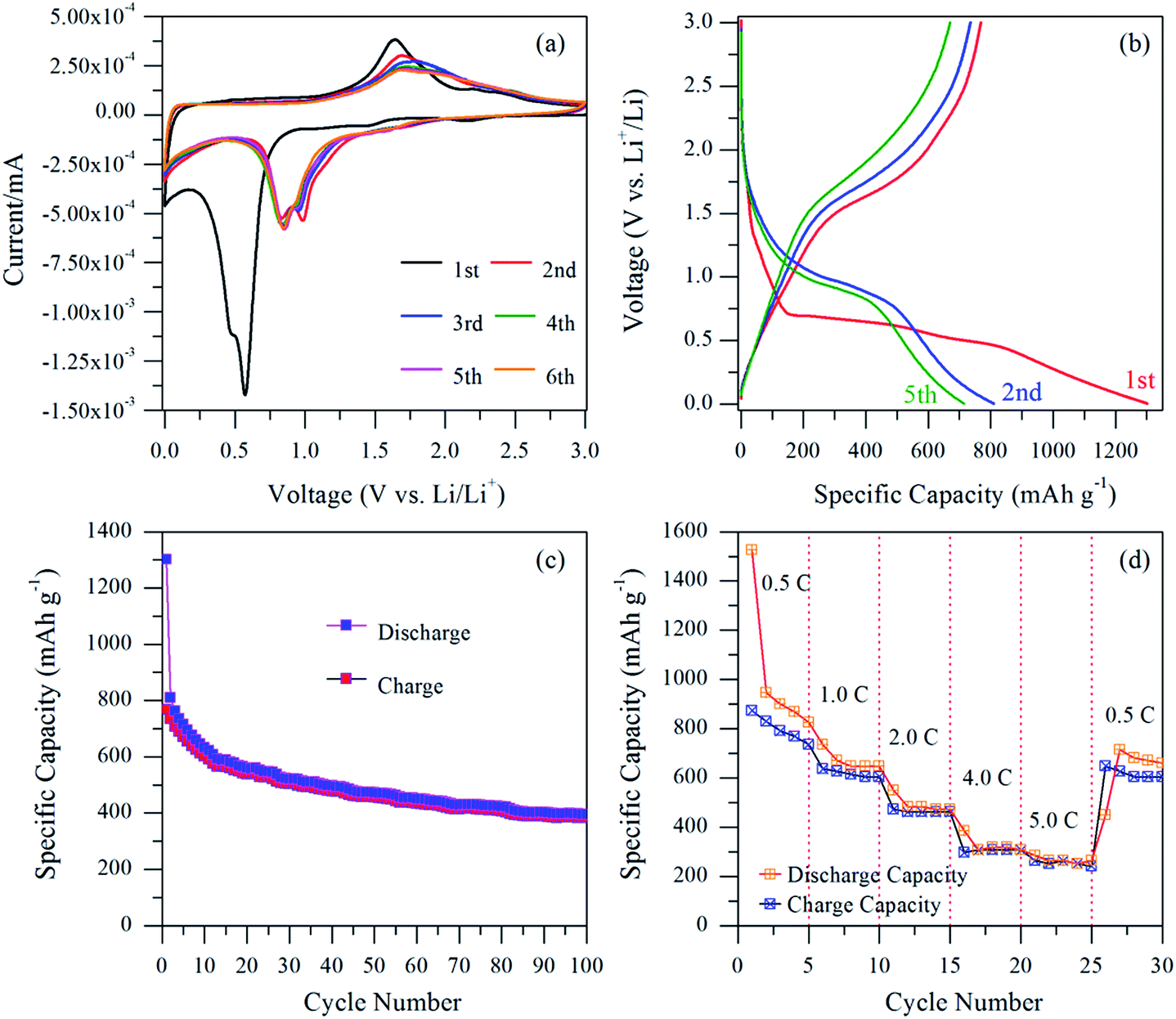 | ||
| Fig. 4 Electrochemical performance of the NiFe2O4/C nanocomposite electrode: (a) cyclic voltammogram at a scan rate of 0.1 mV s−1; (b) discharge/charge voltage profiles at the constant current rate of 1.0C; (c) cycling performance plot at the constant current rate of 1.0C; (d) C-rate capability at various current rates between 0.5C and 5.0C. | ||
Fig. 4(b) shows the galvanostatic charge/discharge profiles for the 1st, 2nd and 5th cycles at a constant current rate of 1.0C [1C = 915 mA g−1] in the voltage range of 0.005 to 3.0 V. The 1st discharge curve consists of two slope regions and a plateau. The first slope between 3.0 V–0.7 V can be attributed to Li+ ion insertion into the electrode. Furthermore, the long flat voltage plateau between 0.7 V–0.45 V, followed by a gradual slope to the cut-off voltage of 0.005 V can be ascribed to the reduction of NiFe2O4 and the formation of Li2O as well as the formation of SEI layers. More precisely, it can also be observed that the large discharge plateau disappears in the later cycles, indicating that irreversible reactions such as electrolyte decomposition or SEI layer formation occurred. The 1st discharge and charge capacities were found to be 1301.7 mA h g−1 and 768.0 mA h g−1, respectively with an initial coulombic efficiency of 59%. The large irreversible capacity loss in the first cycle can be attributed to the polymer gel like SEI layer formation, irreversible decomposition of the electrolyte and some of the undecomposed Li2O phase.10 In the subsequent 2nd and 5th cycles, the discharge and charge capacities are reduced to 808.9 mA h g−1 and 734.7 mA h g−1, and 713.9 mA h g−1 and 670.1 mA h g−1, respectively with a high coulombic efficiency of 91% and 94%. The coulombic efficiency rapidly increases and then remains almost the same in the subsequent cycles. It is believed that the carbon network favours better interparticle-connectivity and hence improves the electronic conductivity of the electrode.
Fig. 4(c) shows the cycling behaviour of the NiFe2O4/C electrode up to 100 cycles at a constant current rate of 1.0C. From the profile, it can be seen that the NiFe2O4/C electrode shows a stable and high reversible capacity except for the initial few cycles. The reversible charge capacity of the NiFe2O4/C electrode is 381.8 mA h g−1 after 100 cycles, which is still higher than the theoretical capacity of the graphite anode (372 mA h g−1). It is highly probable that the good cycling capacity of the electrode is due to the small size of the nanoparticles, which shorten the Li+ ion transport distance and increase the kinetics of the conversion reactions. In addition, the existence of a carbon network imparts excellent electronic conductivity to the electrode material and buffers the volume changes during cycling to decrease pulverization and deterioration of the NiFe2O4 electrode during cycling. In order to clearly evaluate the performance of the pyro-synthesized NiFe2O4/C electrode, a detailed comparison between the current study and previous NiFe2O4 studies based on different synthesis methods, various microstructures and nanocomposites with graphene nanosheets and carbon materials is shown in Table 1.7,10,15,17,22–24 It can be seen that the obtained capacity of the current NiFe2O4/C electrode is better than those reported in the literature. More importantly, the synthesis strategy of the present study is also cost-effective and simple compared to the previous reports.
| Materials | Synthesis method | Current density/rate | Cycle number | Specific capacity (mA h g−1) | Ref. |
|---|---|---|---|---|---|
| NiFe2O4 octahedron | Hydrothermal method | 0.1C | 50 | 375 mA h g−1 | 7 |
| NiFe2O4/C composites | Polymer pyrolysis method | 1/8C | 40 | 780 mA h g−1 | 17 |
| — | — | 4C | 40 | 200 mA h g−1 | — |
| NiFe2O4 mesoporous spheres | Template-free hydrothermal method | 0.2C | 50 | 223 mA h g−1 | 22 |
| NiFe2O4/rGO | Hydrothermal route | 0.2C | 50 | 489 mA h g−1 | 15 |
| Porous core–shell NiFe2O4@TiO2 nanorods | Metal–organic frameworks template | 5.5C | 50 | 202 mA h g−1 | 10 |
| NiFe2O4 nanoparticles | Hydrothermal | 0.2 mA cm−2 | 3 | 709 mA h g−1 | 23 |
| NiFe2O4 nanorods | Template-engaged reaction | 1C | 300 | 520 mA h g−1 | 24 |
| NiFe2O4/C | Pyro-synthesis | 1C | 100 | 381.8 mA h g−1 | Current study |
| NiFe2O4/C | Pyro-synthesis | 5C | 25 | 263.7 mA h g−1 | Current study |
Fig. 4(d) demonstrates the remarkable rate capability of the NiFe2O4/C electrode at various current rates between 0.5C and 5.0C. With the increase of current rate, the capacity decreases. It maintains a reversible charge capacity of 791.2 mA h g−1, 615.4 mA h g−1, 461.5 mA h g−1, 307.7 mA h g−1 and 263.7 mA h g−1 at a current rate of 0.5C, 1.0C, 2.0C, 4.0C and 5.0C, respectively. It can also be seen that a charge capacity of 648.4 mA h g−1 could be recovered when the current density returns to 0.5C. In addition, the NiFe2O4/C electrode exhibits stable capacities at all the high current rates of 1.0C, 2.0C, 4.0C and 5.0C. It is possible that the carbon existing in the system is responsible for the lower charge transfer resistance of the hybrid material.
In order to further confirm that the NiFe2O4 nanoparticles are protected by the carbon network during cycling, an ex situ TEM study is performed on the cycled electrode after 60 cycles. Briefly, for the ex situ TEM studies, the electrode was separated from the coin cell inside the glove box and subsequently washed thoroughly with solvent (dimethyl carbonate) to remove the electrolyte. Fig. 5(a) and (b) show the ex situ FE-TEM images of the NiFe2O4/C electrode after 60 cycles at different magnifications. It can be clearly seen that the carbon network and electrode integrity is still maintained after long term cycling. The NiFe2O4 nanoparticles are still completely anchored on the carbon network, ensuring good electrical contact between the NiFe2O4 nanoparticles and carbon. Finally, it is also reasonable to suggest that the small NiFe2O4 nanoparticles anchored on the carbon network could alleviate the pulverization problem and enhance the electrical conductivity, cycling performance and rate capabilities of the NiFe2O4/C electrode.
 | ||
| Fig. 5 Ex situ FE-TEM images of the NiFe2O4/C nanocomposite electrode at different magnifications after 60 cycles of discharging/charging at a constant current rate of 1.0C. | ||
4. Conclusions
In summary, an easy, fast and low cost pyro-synthesis approach was chosen to fabricate NiFe2O4 nanoparticles decorated onto a conductive carbon network in order to overcome the issues of NiFe2O4 such as large volume changes and poor electrical conductivity. The material was systematically characterized using X-ray diffraction, field-emission scanning electron microscopy, field-emission transmission electron microscopy, Raman spectroscopy and electrochemical measurements. Morphology observation clearly shows that the NiFe2O4 nanoparticles are well anchored within the carbon network with particle sizes of 50–100 nm. As an anode, the NiFe2O4/C electrode exhibits a high reversible capacity of 381.8 mA h g−1 after 100 cycles at a constant current rate of 1.0C and a better rate capability of 263.7 mA h g−1 at 5.0C. It is believed that the carbon network in this architecture is not only providing the electrical network to facilitate Li+ ion and electron transport, but also offers buffering during electrochemical cycling to maintain the structural integrity of the electrode. In addition, it may also be probable that the presence of the extra phase of Fe2O3 in the nanocomposite sample may also contribute to improving the electrochemical performance of the parent NiFe2O4/C electrode by the storage of six Li+ ions per formula unit. The current strategy can be extended to produce other hybrid metal oxides with novel architectures for high-performance energy storage applications.Acknowledgements
A. K. Rai is grateful for the financial support by the Science and Engineering Research Board (SERB), Government of India, vide grant no. YSS/2015/000489. We are also thankful to the Amrita Centre for Nanosciences for providing the infrastructure.References
- T. V. Thi, A. K. Rai, J. Gim and J. Kim, J. Power Sources, 2015, 292, 23 CrossRef CAS.
- L. Huang, G. H. Waller, Y. Ding, D. Chen, D. Ding, P. Xi, Z. L. Wang and M. Liu, Nano Energy, 2015, 11, 64 CrossRef CAS.
- Y. N. NuLi and Q. Z. Qin, J. Power Sources, 2005, 142, 292 CrossRef CAS.
- L. Yao, X. Hou, S. Hu, J. Wang, M. Li, C. Su, M. O. Tade, Z. Shao and X. Liu, J. Power Sources, 2014, 258, 305 CrossRef CAS.
- A. K. Rai, S. Kim, J. Gim, M. H. Alfaruqi, V. Mathew and J. Kim, RSC Adv., 2014, 4, 47087 RSC.
- L. Lu, J. Z. Wang, X. B. Zhu, X. W. Gao and H. K. Liu, J. Power Sources, 2011, 196, 7025 CrossRef CAS.
- L. Liu, L. Sun, J. Liu, X. Xiao, Z. Hu, X. Cao, B. Wang and X. Liu, Int. J. Hydrogen Energy, 2014, 39, 11258 CrossRef CAS.
- M. H. Alfaruqi, A. K. Rai, V. Mathew, J. Jo and J. Kim, Electrochim. Acta, 2015, 151, 558 CrossRef CAS.
- C. T. Cherian, J. Sundaramurthy, M. V. Reddy, P. S. Kumar, K. Mani, D. Pliszka, C. H. Sow, S. Ramakrishna and B. V. R. Chowdari, ACS Appl. Mater. Interfaces, 2013, 5, 9957 CAS.
- G. Huang, F. Zhang, X. Du, J. Wang, D. Yin and L. Wang, Chem.–Eur. J., 2014, 20, 11214 CrossRef CAS PubMed.
- H. Liu, H. Zhu and H. Yang, Mater. Res. Bull., 2013, 48, 1587 CrossRef CAS.
- P. Lavela, N. A. Kyeremateng and J. L. Tirado, Mater. Chem. Phys., 2010, 124, 102 CrossRef CAS.
- C. T. Cherian, M. V. Reddy, G. V. Subba Rao, C. H. Sow and B. V. R. Chowdari, J. Solid State Electrochem., 2012, 16, 1823 CrossRef CAS.
- Y. Xiao, J. Zai, X. Li, Y. Gong, B. Li, Q. Han and X. Qian, Nano Energy, 2014, 6, 51 CrossRef CAS.
- P. Zhu, S. Liu, J. Xie, S. Zhang, G. Cao and X. Zhao, J. Mater. Sci. Technol., 2014, 30, 1078 CAS.
- A. K. Rai, T. V. Thi, B. J. Paul and J. Kim, Electrochim. Acta, 2014, 146, 577 CrossRef CAS.
- Y. Ding, Y. Yang and H. Shao, J. Power Sources, 2013, 244, 610 CrossRef CAS.
- J. Kang, S. Baek, V. Mathew, J. Gim, J. Song, H. Park, E. Chae, A. K. Rai and J. Kim, J. Mater. Chem., 2012, 22, 20857 RSC.
- A. K. Rai, J. Jim, T. V. Thi, D. Ahn, S. J. Cho and J. Kim, J. Phys. Chem. C, 2014, 118, 11234 CAS.
- Z. Li, Y. Wang, H. Sun, W. Wu, M. Liu, J. Zhou, G. Wu and M. Wu, J. Mater. Chem. A, 2015, 3, 16057 CAS.
- J. Yang, J. Wang, X. Li, D. Wang, J. Liu, G. Liang, M. Gauthier, Y. Li, D. Geng, R. Li and X. Sun, J. Mater. Chem., 2012, 22, 7537 RSC.
- L. Duan, Y. Wang, L. Wang, F. Zhang and L. Wang, Mater. Res. Bull., 2014, 61, 195 CrossRef.
- H. Zhao, Z. Zheng, K. W. Wong, S. Wang, B. Huang and D. Li, Electrochem. Commun., 2007, 9, 2606 CrossRef CAS.
- N. Wang, H. Xu, L. Chen, X. Gu, J. Yang and Y. Qian, J. Power Sources, 2014, 247, 163 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |