
Mark–Houwink–Sakurada coefficients determination for molar mass of silk fibroin from viscometric results. SEC-MALLS approach
Abstract
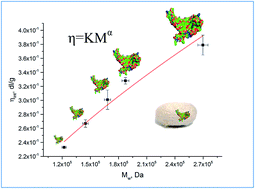
The results on the changes of molar mass distributions and average molar masses in Bombyx mori fibroin with use of size exclusion chromatography (SEC) and viscometry, respectively, are presented in terms of the determination of Mark–Houwink–Sakurada coefficients, which are lacking in the literature. SEC was applied in configuration of light scattering and differential refractive index and multi angle laser light scattering detectors (SEC-DRI-MALLS). The coefficients calculated from the correlation between intrinsic viscosities and the absolute value of molar masses obtained using the SEC-MALLS technique are equal to: K = 1.813 × 10−4 dm3 g−1 (±1.74 × 10−5), α = 0.614 (±0.068). The value of the parameter α shows that silk fibroin molecules in an LiBr solution behave like a flexible polymer molecule in a thermodynamically good solvent. The results are discussed with focus on the applicability of glass capillary viscometry and size exclusion chromatography for assessment of silk based materials condition with particular reference to the preservation artifacts and ancient textiles.
 Please wait while we load your content...
Please wait while we load your content...

