
Self-supported three-dimensionally interconnected polypyrrole nanotubes and nanowires for highly sensitive chemiresistive gas sensing
Abstract
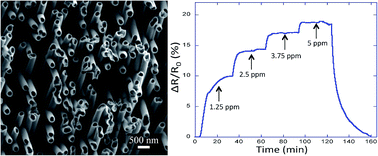
We developed a versatile template-based fabrication method for growing large arrays of three-dimensionally interconnected polypyrrole nanotubes (or nanowires) with easily tunable geometrical dimensions and spatial arrangement. Such a macroscopic network made up of conducting polymer elongated nanostructures provides an extremely large active surface with increased electrical connectivity, as well as an enhanced structural integrity of the flexible network. The three-dimensional array of polypyrrole nanofibers exhibited an excellent sensitivity towards gaseous ammonia, providing reliable and accurate detection at gas concentrations as low as 1 ppm. The novel preparation approach offers a cost-effective alternative for large-scale production of easily integrable chemiresistive sensors for different applications.
 Please wait while we load your content...
Please wait while we load your content...

