
Metallic glass formation in Cu–Ni–Ti (Zr, Hf) systems studied by thermodynamic calculations
Abstract
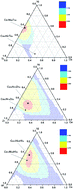
For the family of Cu–Ni–Ti (Zr, Hf) systems, which are promising for obtaining bulk metallic glasses, glass formation regions were calculated based on the extended Miedema’s model and Alonso’s method. It is found that the calculated glass formation regions of the Cu–Ni–Zr and Cu–Ni–Hf systems agree well with experimental results, whereas it does not for the Cu–Ni–Ti system. The composition dependence of the glass forming ability in the Cu–Ni–Ti (Zr, Hf) systems were then predicted, and it turns out that the glass forming ability of the Cu–Ni–Ti system largely deviates from the experimental results, from which it is assumed that kinetic factors (low liquidus temperature) instead of thermodynamic factors cause the Cu-rich composition to easily form glass in the Cu–Ni–Ti system. Meanwhile, the effect of Ti (Zr, Hf) on the glass forming ability was discussed in terms of the mixing enthalpy and atomic size effect.
 Please wait while we load your content...
Please wait while we load your content...

