DOI:
10.1039/C6RA03128E
(Paper)
RSC Adv., 2016,
6, 37610-37620
Preparation, characterization and in vitro photodynamic therapy of a pyropheophorbide-a-conjugated Fe3O4 multifunctional magnetofluorescence photosensitizer
Received
2nd February 2016
, Accepted 18th March 2016
First published on 21st March 2016
Abstract
Novel pyropheophorbide-a-conjugated multifunctional magnetofluorescence nanoparticles Fe3O4@SiO2@APTES@Glutaryl-PPa (MFNPs) with a mean diameter of 50 nm were strategically designed and prepared for photodynamic therapy (PDT) and medical fluorescence imaging. Chlorin photosensitizer pyropheophorbide-a (PPa) was covalently anchored on the surface of core–shell Fe3O4@SiO2@APTES nanoparticles that were prepared via a sol–gel process with a bridging glutaryl group. The phase constitution, morphology, size, chemical properties, magnetic property of the intermediates and final nanoparticles were characterized by X-ray powder diffraction, transmission electron microscopy, Fourier transform infrared spectrometer, zeta potential, vibration sample magnetometer, thermogravimetric analysis, ultraviolet-visible absorption spectra and fluorescent emission spectroscopy. These results showed that the MFNPs have good dispersibility in alcohol and water, excellent magnetization with 17.31 emu g−1 at 300 K, strong superparamagnetic and good photoluminescence property. The in vitro PDT against the human HeLa cervical cancer cell suggested that MFNPs could permeate the tumor cells quickly and possess suitable lipo-hydro partition coefficient, inducing damage and apoptotic cell death. The cancer cell viability was lowered to 10.18% after treatment with PDT. In addition, the formation of reactive oxygen species in HeLa cells after MFNPs-PDT treatment was studied, which suggested that Type I and Type II photodynamic reactions can occur simultaneously.
Introduction
Cancer has been seriously threatening human health for a long period. Among the various choices for cancer treatment, photodynamic therapy (PDT),1–3 as a noninvasive therapeutic modality providing painless and repeating treatment for patients, has obtained regulatory approval for clinical applications for various kinds maladies such as gastric cancer,4 urinary system tumors,5 breast cancer,6 brain tumors, actinic keratoses and psoriasis,7,8 etc. PDT involves the combination of visible light, photosensitizer, and tissue oxygen. In the presence of oxygen, the tumor-associated photosensitizer is excited from ground state to the excited state after activation with light of an appropriate wavelength, and an electron was transferred to nearby tissue oxygen, producing oxygen free radicals (Type I reaction mechanism of PDT), or hydroxyl radicals (HR), superoxide anion, hydrogen peroxide and so on; or the excitation energy might then be transferred to nearby tissue oxygen, thus producing excited singlet oxygen (1O2, Type II reaction mechanism of PDT). In spite of their short-time existence, these oxygen free radicals and singlet oxygen, also known as Reactive Oxygen Species (ROS),9,10 have a very strong activity to induce the cell toxicity or destroy the cells, leading to vascular injury or abnormal immune response, and eventually leading to cancer tissue damage.
Photosensitizer is the key of PDT. The ideal photosensitizer should not only be able to specially bind to tumor cells, have a strong absorption at a wavelength of 600–900 nm region, high efficiency in generating reactive oxygen species within the aerobic tissues, but also have characteristics of cancer targeting, low dark toxicity, soft tissue penetration depth, and so on. Natural chlorophyll and its degradation product pyropheophorbide-a (PPa) are potential ideal materials for photodynamic therapy due to their advantages of long absorption wavelength (>667 nm), low dark toxicity, well-defined composition and molecular structure, big molar extinction coefficient and high rate of fluorescence quantum.11 Nevertheless, because of the π–π conjugation between the parent nucleus chlorin aromatic ring, PPa is prone to aggregation under physiological conditions, leading to the low degree of natural selective aggregation in the lesion tissue, low water solubility and poor target enrichment of PPa, increasing the poisonous side effect on the organizational system. Herein, improving the water solubility of pyropheophorbide-a, reducing the aggregation under physiological conditions and enhancing selective enrichment in diseased tissue, is the hinges to develop pyropheophorbide-a as an ideal photosensitizer in PDT in clinical application.
In recent years, magnetic Fe3O4 NPs, which have the advantages of good magnetic targeting, surface effect and small-sized effect and biocompatibility, have been widely applied in fields of targeted drug deliver, cell separation,12 magnetic resonance imaging (MRI),13–17 biosensing,18 immunoassay,19,20 catalytic,21 and magnetic hyperthermia.22,23 Superparamagnetic Fe3O4 are preferred due to its ability to become magnetized upon exposure to a magnetic field but have no permanent magnetization once the field is turned off.24 Thus, they are less prone to aggregate and to induce dangerous thrombosis of blood vessels. So it's a potential reagent for targeted drug carrier in drug delivery applications. While pure Fe3O4 NPs were easy to aggregate because of the nano-effect and magnetic attraction, and easy to be oxidized in the air, which has limited the direct application.25,26 Coating SiO2 layer on the surface of Fe3O4 has been widely recognized for which could effectively reduce the zero charge points, shield the magnetic attraction and transfer the phase from hydrophobic to hydrophilic, consequently leading to better water-solubility, chemical stability and biocompatibility.27,28 In addition, the abundant hydroxyl or amino groups on the surface of SiO2 make the particles easy to further functionalize.29–31 A recently reported Fe3O4@TiO2 NPs could be used as a multifunctional agent of MRI and an inorganic photosensitizer for PDT.32 TiO2 could be maintained for a long time in the body than that of organic photosensitizers, yet the poor water-solubility, short absorption wavelength only response to ultraviolet light and soft tissue penetration shallow had limited its application for PDT. Another newly reported magneto-fluorescence organic photosensitizer chlorin e6 (Ce6) conjugated Fe3O4 is suitable for simultaneous targeting PDT and in vivo MRI,4 yet with poor dispersity and a short absorption wavelength lower than 650 nm. The designed photosensitizer in our paper showed longer absorption wavelength (667 nm) than the above reported agents. The longer absorption wavelength makes the photosensitizer more potential in the treatment of deep tumors.
Herein, we designed and synthesized a novel multifunctional magneto-fluorescence nanoparticles Fe3O4@SiO2@APTES@Glutaryl-PPa (MFNPS) (Fig. 1) through a bridging glutaryl group for simultaneous photodynamic therapy, medical fluorescence imaging, magnetic hyperthermia and noninvasive MRI.4,33 In this study, Fe3O4 was first coated with SiO2 layer, which was used to make the particles easily functionalized, and then 3-aminopropyl triethoxysilane (APTES) was conjugated to the surface of iron oxide nanoparticles through hydroxyl group, so that the amino groups on the surface of nano-Fe3O4 could make the particles easy to couple with PPa through glutaryl dichloride coupling agent and improve the dispersion of MFNPS in water or alcohol. Final products MFNPS simultaneously possessed photodynamic therapy activity and good magnetic targeting. Mostly important, the core–shell Fe3O4@SiO2@APTES and glutaryl group will significantly improve the dispersity and biocompatibility, enhance the tumor targeting, reduce toxicity, and overcome drug-resistance mechanisms. The target MFNPs was synthesized through continuous chemical modifications on nano-Fe3O4 core (Fig. 1). The properties of the intermediates and product were characterized using various methods. The in vitro PDT against human HeLa cervical cancer cell line was investigated to evaluate MFNPs as the photosensitizer agent in support of PDT. In order to investigate the intracellular distribution of MFNPs, cell uptake experiments were performed on Hela cells and analyzed by fluorescent inverted microscope. Cell morphological changes after PDT was analyzed by using acridine orange (AO)/ethidium bromide (EB) double fluorescent staining. In addition, specific ROS quenching agent SA and DM were utilized to visualize Type I and Type II photodynamic reactions. The research provided a unique method of the preparation of different MFNPs by engineering the structure of Fe3O4 core and expand the application of multifunctional nanoparticles in the field of PDT, fluorescence imaging, magnetic hyperthermia and MRI.
 |
| | Fig. 1 Schematic diagram of the synthesis route for the magneto-fluorescence nanoparticles MFNPs (Fe3O4@SiO2@APTES@Glutaryl-PPa) with core–shell structure. | |
Experimental
Chemicals and reagents
Ferric chloride hexahydrate (FeCl3·6H2O, 98%), sodium oleate, oleic acid, tetraethyloxylsilicate (TEOS), hexadecyl trimethyl ammonium bromide (CTAB, 99%), (3-aminopropyl)-triethoxy silane (APTES), dimethyl sulfoxide (DMSO) and 3-(4,5-dimethy lthiazol-2-yl)-2,5-dipheny-ltetrazolium bromide (MTT) were purchased from Sigma-Aldrich. 1-Octadecene, glutaryl dichloride were purchased from Alfa Aesar. Chemical Company. Ethanol, toluene, chloroform, triethanolamine, methanol, dichloromethane, sodium hydroxide (NaOH), were analytical reagent. Dulbecco's modified eagle medium (DMEM), penicillin, fetal bovine serum (FBS), and streptomycin were purchased from Beijing Dingguo Biotechnology Co. Phosphate buffered saline (PBS) purchased from Invitrogen (10010) was used as a balanced salt solution in cell culture. PBS used in other experiments was prepared by mixing stock solutions of NaH2PO4 and Na2HPO4. All the above chemicals reagents were used without further purification. All the solvents were distilled and purified by standard procedures. The pure water was obtained from a Milli-Q synthesis system (Millipore, Billerica, MA, USA).
Characterization
The size and shape of the nanoparticles were observed by a Tecnai G2 F20 S-TWIN transmission electron microscope (TEM) (FEI, America) operating at 200 kV. The X-ray powder diffraction (XRD) analysis was performed using a Dmax-2600/PC (Rigaku Corporation, Tokyo, Japan) in the range of 20–80° with a rate of 3° min−1. Cu Kα radiation (λ = 1.5406 Å) was used and the tube operated at 40 kV and 30 mA. Fourier transform infrared (FT-IR) spectra were acquired using Vertex 80 FTIR spectrometer (Bruker Co., German) with a resolution of 4 cm−1 as KBr disc in the range of 4000–400 cm−1. Magnetic characteristics were performed at 300 K using a 7410 vibrating sample magnetometer (VSM) (Lake Shore, America). The thermal analysis (TGA) was performed using Diamond 6300 TGA (Diamond, America). Analysed sample was heated from 25 to 800 °C at a heating rate of 10 °C min−1 under a nitrogen flow of 50 mL min−1. UV-vis absorption spectra were measured on LAMBDA 25 spectrometer (PerkinElmer). Fluorescent emission spectroscopy was measured on DF-1000 (China). Zeta potential was carried out with NanoBrook ZetaPALS Zeta Potential Analyzer (Brookhaven). Biotek ELx800 absorbance microplate reader was used in MTT assay. The cells were imaged using a Fluorescent inverted microscope (FIM, Leica DM IL LED, Leica Microsystems, Germany).
Synthesis of magnetic Fe3O4 NPs
Ferric chloride hexahydrate (FeCl3·6H2O 2.7 g, 10 mmol) and sodium oleate (9.13 g, 30 mmol) were dissolved in a mixture solvent (20 mL EtOH, 15 mL distilled water, 35 mL hexane), ultrasonically for 30 min. The mixed solution was stirred at 70 °C for 4 h, and then cooled to room temperature. The upper organic layer which contained the iron–oleate complex was separated in a separation funnel and washed with 10 mL distilled waters. After evaporating off the organic solvent, iron–oleate complex was obtained in a waxy solid form.34,35 Afterward, iron–oleate complex (9 g, 10 mmol) was dissolved in octadecene (50 g, 198.4 mmol) and oleic acid (1.425 g, 4.685 mmol). After 10 min of ultrasonic dispersion at room temperature, the reaction mixture was heated to 320 °C (3.3 °C min−1) and kept this temperature for 30 min. The color of the transparent reaction mixture became turbid and brownish black. After cooling to room temperature, added 125 mL of anhydrous ethanol to precipitate the nanocrystals. In high speed centrifugation condition (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm), Fe3O4 nanoparticles were separated and then dumped to remove the upper layer. 400 mL of mixture solvent (volume ratio, V, ethanol:chloroform) was added several times and separate the Fe3O4 by high speed centrifugation again. Collected black Fe3O4 and vacuum dried at 60 °C, then dispersed in chloroform for further use.
000 rpm), Fe3O4 nanoparticles were separated and then dumped to remove the upper layer. 400 mL of mixture solvent (volume ratio, V, ethanol:chloroform) was added several times and separate the Fe3O4 by high speed centrifugation again. Collected black Fe3O4 and vacuum dried at 60 °C, then dispersed in chloroform for further use.
Synthesis of Fe3O4@SiO2 NPs
The core–shell Fe3O4@SiO2 NPs were prepared by employing sol–gel method and tetraethyl orthosilicate (TEOS) as precursor under alkaline conditions and then incorporated with the proper amount of surfactant, cetyltrimethyl ammonium bromide (CTAB).25 In a typical procedure, 25 mg Fe3O4 NPs were dispersed in 45 mL of chloroform, ultrasonically for 1 h. Then pouring Fe3O4 into the solution which contained 3 g hexadecyl trimethyl ammonium bromide and 80 mL of deionized water. The mixture solution was stirred vigorously on a water bath for 30 min at 32 °C. Next, the mixture was maintained heated up to 60 °C to evaporate the chloroform. After stirring 30 min at 32 °C, NaOH aqueous solution (0.1 M) was added into the system to adjust the pH between 8 and 9. 550 μL of 20% TEOS ethanol solution was added several times. And stirred vigorously for 24 h at this temperature. After adding 100 mL of ethanol, stirred vigorously and then the Fe3O4@SiO2 were collected by centrifugation and washed the black solid with little ethyl alcohol three times. Then dispersed in 50 mL ethanol for further use.
Synthesis of Fe3O4@SiO2@APTES NPs
The Fe3O4@SiO2@APTES microspheres were synthesized by the reaction between (3-aminopropyl)-triethoxysilane (APTES) and hydroxyl groups. Briefly, 45 mg Fe3O4@SiO2 NPs were scattered in methylbenzene (60 mL) containing 260 μL of APTES, and the mixture was stirred vigorously for 7 h. After cooling to room temperature. The product Fe3O4@SiO2@APTES NPs was separated by an external magnet and washed with 100 mL ethanol three times by centrifugation. Then, dried in a vacuum oven at 50 °C for 3 h and kept it under the condition of cutting off from the air.
Synthesis of Fe3O4@SiO2@APTES@Glutaryl-PPa (MFNPS) magne-to-fluorescence microspheres via bridging glutaryl group
Glutaryl dichloride (22 μL, 0.27 mmol) and triethylamine (72 μL, 0.81 mmol) were dissolved in dried CH2Cl2 (15 mL). Under the condition of stirring, a mixture which contained 100 mg PPa and dried CH2Cl2 (20 mL) was dripped into the system slowly. After dripping, stirred vigorously for 2 h. During the above reaction process, 10 mg Fe3O4@SiO2@APTES NPs were dispersed in dried CH2Cl2 (40 mL), ultrasonically for 1 h. Then, 72 μL of triethylamine was added to the reaction system. The mixture which contained Fe3O4@SiO2@APTES and dried CH2Cl2 was dripped into the system slowly. After dripping, stirred vigorously overnight. Then, the precipitated nanoparticles were separated by an external permanent magnet and the liquid was pumped to remove. Washed the solid which were adsorbed by a magnet with 20 mL of absolute methanol several times, dried in a vacuum oven at 50 °C for 3 h, and kept it airproof in dry and cool place and kept away from light.
Cell culture
The human cervical cancer cells line (HeLa) were maintained in cell culture dishes with Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% antibiotic (100 μg mL−1 penicillin to 100 μg mL−1 streptomycin, Life Technologies, USA) in an incubator containing 5% CO2 and 98% humidity at 37 °C, and culture media were changed as needed, and cells were passaged every other day.
MTT colorimetric assay
To check the cytotoxicity of the MFNPs, cell viability was estimated by means of the colorimetric MTT assay. The cells adhered to the surface of the culture vessel were dissected into a cell suspension by using 0.25% trypsin digestion. Followed, 200 μL of 105 cell per millilitre suspension was inoculated into a 96-well plate and cultured as described above. Cells in experimental groups were cultured with different concentrations of MFNPs (each concentration gradient) for 3 h followed by exposure to calibrated visible light from a Xenon Lamp (Newport 67005 Oriel Instruments) passed through a 675 nm filter (FSQ-GG400) for 10 min (dosage 25 J cm−2) and culture in dark for additional 24 h. Then an MTT (Sigma) solution in PBS (20 μL, 5 mg mL−1) was added to each well followed by incubation for 4 h under the same environment. The culture medium was then aspirated and 150 μL dimethyl sulfoxide was added to dissolve the sediment. The absorbance was then measured with a Biotek ELx800 absorbance microplate reader at a wavelength of 490 nm. Cell viability (%) was then calculated by the equation:
| Cell viability (%) = Ã 490(sample)/Ã 490(control) × 100% |
where à 490(sample) is the average absorbance of the wells treated with various concentrations of MFNPs, and à 490(control) is the average absorbance of the wells treated with DMEM + 10% FBS. Each experiment was repeated six times.36
Cellular uptake of MFNPs nanoparticles
The in vitro uptake of the magneto-fluorescence nanoparticles MFNPs in Hela cancer cell was analyzed by fluorescent inverted microscope (FIM). Suspensions of 105 cell per millilitre of the HeLa cell lines were incubated on 6 well plates at a density of 2 × 105 cells per well and the plates were covered with autoclaved cover glass, followed by incubation overnight. The MFNPs (1 mL, 60 μg mL−1) was added to the cultured cell in each well, incubated for 0.5, 1, and 3 h respectively, and subsequently rinsed with cold PBS three times. Then, cells were fixed with glutaraldehyde solution (1 mL, 2.5%) for 10 min at 37 °C, the glutaraldehyde solution was removed and the cells extensively rinsed with cold PBS three times again, and subsequently stained with 1 mL of a 1 μg mL−1 DAPI nuclear probe for 10 min at room temperature. Cell imaging was performed on a Leica DM IL LED fluorescent inverted microscope (FIM).37,38
Morphological changes of HeLa cells after PDT. Cell morphological changes were analyzed by AO (acridine orange)/EB (ethidium bromide) double fluorescent staining, which was used to label the nuclear DNA. Suspensions of 105 cell per millilitre of the HeLa cell lines were incubated on 6 well plates at a density of 2 × 105 cells per well and incubated 24 h. The MFNPs (1 mL, 50 μg mL−1) was added to each well with irradiation for 10 min, and subsequently incubated for 6 h. After removing the culture medium, AO/EB (5 μL, 100 μg mL−1) dye solutions were added to the cultured cell in each well. Then morphological variation was observed by fluorescent inverted microscope.
In vitro PDT effect and cell viability MTT assays
The photodynamic activity test was divided into two groups: experimental groups, with cells treated with different concentrations of MFNPs and exposed to light; dark control group keeps identical to the experimental group without irradiation. About 2 × 105 cells per well were seeded into 96-well plates and incubated for 24 h prepared for cytotoxicity assessment. Then the cells of experimental groups were rinsed with PBS, and subsequently cultured with different concentrations of MFNPs (5, 10, 20, 30, 40, 50, 60, 120, 250 μg mL−1, total volume 100 μL per well) for 3 h followed by exposure to calibrated visible light for 10 min (dosage 25 J cm−2), and then cultured in dark for additional 24 h in DMEM media as described above, 5% CO2 and 98% humidity at 37 °C. Cell viability was determined by MTT assay.
Quenching of active oxygen on PDT
To visualize the photochemical processes (Type I and Type II) mechanism of PDT by adding respectively quenching agent. The quencher of sodium azide (SA, Sigma-Aldrich) and D-mannitol (DM, Sigma-Aldrich), which has of relative specificity to singlet oxygen (1O2) and hydroxyl radicals (HR), can quench corresponding ROS generated from photodynamic reaction, respectively.39–41 Thus, the photodynamic activity test was divided into four groups: blank control group without MFNPs and light; MFNPs-PDT group, different concentrations of MFNPs and exposed to light; MFNPs-PDT-SA group, MFNPs with SA and exposed to light; MFNPs-PDT-DM group, MFNPs with DM and exposed to light. Precisely, Hela cells were seeded into 96-well plates at a cell density of 1 × 104 cells per well in DMEM and incubated 24 h as described above. After removing the culture medium and washing with PBS three times, 100 μL of different concentrations of MFNPs was added to each well of 96-well plates of MFNPs-PDT group, MFNPs-PDT-SA group, and MFNPs-PDT-DM group. Besides, SA (20 μL, 1 mol L−1) and DM (40 μL, 1 mol L−1) aqueous solutions were added to each well of MFNPs-PDT-SA group and MFNPs-PDT-DM group, respectively. The final concentrations of MFNPs were 5, 10, 20, 30, 40, 50, 60, 120, 250 μg mL−1. Then the cells in experimental groups were incubated for 4 h followed by exposure to calibrate visible light for 10 min, and then cultured in darkness for additional 24 h in DMEM media at 37 °C under 5% CO2 conditions. While, the cells in the blank control group were treated with DMEM medium at the same concentrations found in the formulations without irradiation. Cell viability was determined by MTT assay.
Results and discussion
Synthesis pathway of PPa-conjugated MFNPs
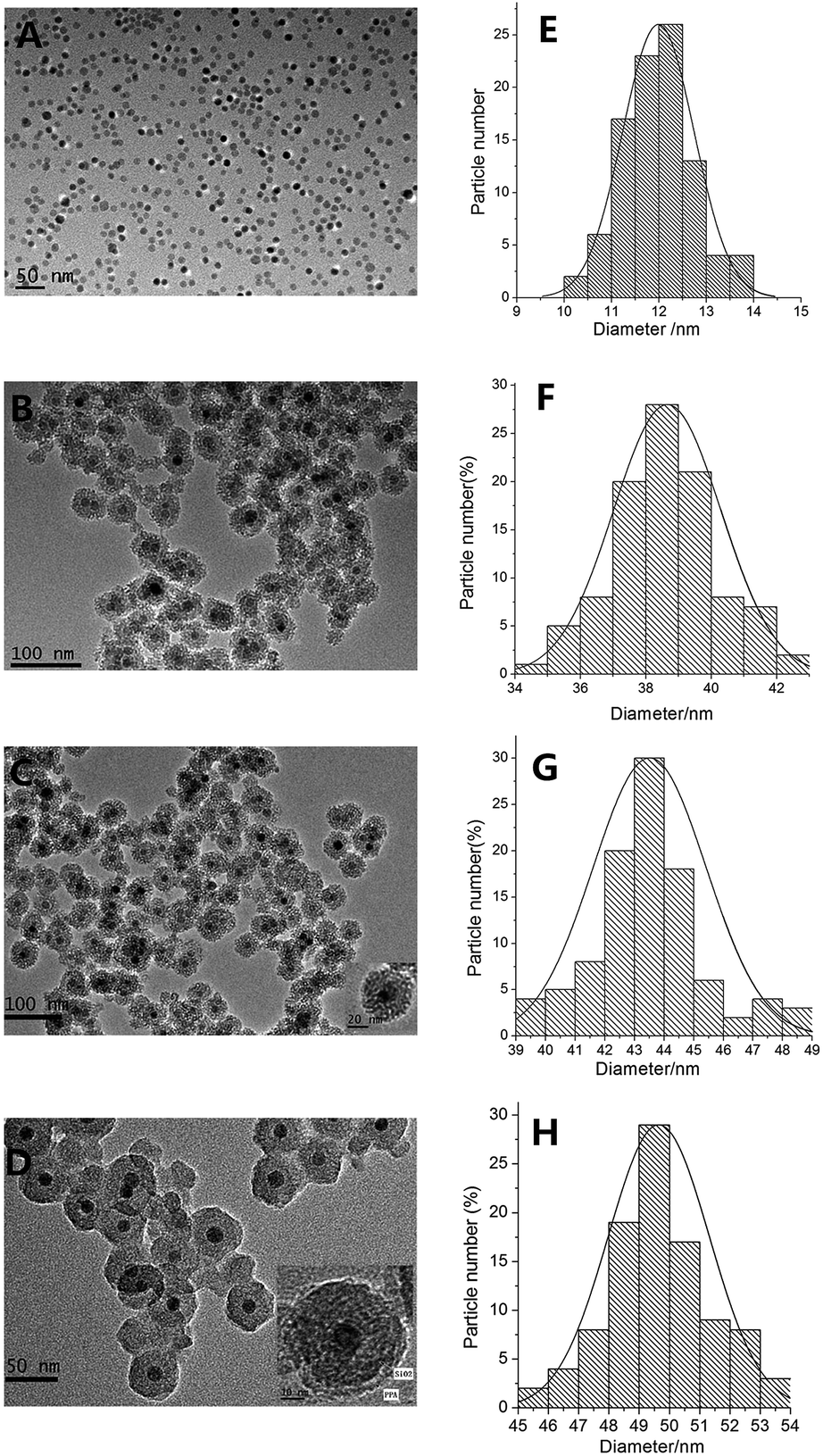
Fig. 1 shows the synthesis of magneto-fluorescence multifunctional nanoparticles. The magnetic Fe3O4 nanoparticles were prepared by the solvothermal method with an average diameter of 12 nm, the core–shell Fe3O4@SiO2, which possesses good dispersity in alcohol, water, chloroform and dichloromethane, were prepared via a sol–gel process with an average diameter of 40 nm, then the obtained nanoparticles were modified by coupling agent APTES to convert to Fe3O4@SiO2@APTES, the amino groups on the surface of SiO2 make the particles easy to further functionalize, finally, Fe3O4@SiO2@APTES@Glutaryl-PPa (MFNPS) with an average diameter 50 nm was conducted with Fe3O4@SiO2@APTES nanoparticles using glutaryl chloride as binding group and triethylamine as alkaline catalyst. Fe3O4@SiO2@APTES@Glutaryl-PPa, a triple-layered core–shell structure magneto-fluorescence nanoparticles, which possesses good dispersity in alcohol and water, excellent magnetization with 17.31 emu g−1 at 300 K. The disperse system was shown in green after being excited by visible light, and red fluorescence under ultraviolet illumination. Due to the coated SiO2 and PPa layer, the magneto-fluorescence nanoparticles were stable in air and acid–base system.
Structure and morphology of MFNPs
Phase constitution of the samples was carried out by X-ray powder diffraction (XRD) technique. Fig. 2 shows XRD patterns of pure Fe3O4 (a), Fe3O4@SiO2 (b), Fe3O4@SiO2@APTES (c), and Fe3O4@SiO2@APTES@Glutaryl-PPa MFNPs (d), respectively. As shown in Fig. 2, the characteristic peaks at 2θ = 30.1°(220), 35.4°(311), 43.1°(400), 53.4°(422), 57.1°(511) and 62.6°(440), which match the magnetite (Fe3O4) crystal structure data well (JCPDS card no. 19-0629). Synthetic Fe3O4 nanocrystalline particles were magnetite crystals with inverse spinel structure. The characteristic peaks and indices were also observed for Fe3O4@SiO2 (b), Fe3O4@SiO2@APTES (c), and Fe3O4@SiO2@APTES@Glutaryl-PPa MFNPs (d), indicating the surface modification and conjugation of Fe3O4 NPs do not change their phase structure. Fig. 2b displays the XRD pattern of Fe3O4@SiO2 showing an obvious broad peak at 2θ = 15–30°, which is generally considered as the diffusion peak of amorphous silica. Fig. 2c displays the XRD pattern of Fe3O4@SiO2@APTES, the diffusion peak of amorphous silica at 2θ = 15–30° is broader and higher than that in Fig. 2b, indicating that APTES is successfully coated on the surface of Fe3O4@SiO2. Fig. 2d displays the XRD pattern of Fe3O4@SiO2@APTES@Glutaryl-PPa MFNPs, the diffusion peak of amorphous silica at 2θ = 15–30° is much broader and higher than that in Fig. 2c, moreover, the peak type has a tendency to become sharper, showing that the chlorin photosensitizer (PPa) has a certain effect on the diffusion peak of amorphous silica, suggesting further that PPa has been coated on the surface of Fe3O4@SiO2@APTES nanoparticles successfully through glutaryl group. These results confirm that the nanoparticles synthesized in this study are the Fe3O4.
 |
| | Fig. 2 X-ray diffraction (XRD) patterns of Fe3O4 (a), Fe3O4@SiO2 (b), Fe3O4@SiO2@APTES (c), and Fe3O4@SiO2@APTES@Glutaryl-PPa (d), respectively. | |
The morphology and sizes of samples were investigated by transmission electron microscopy (TEM) in part A–D of Fig. 3, and the size distribution statistical analysis from the TEM were presented in parts E–H of Fig. 3. The mean sizes of Fe3O4 NPs, Fe3O4@SiO2 NPs, Fe3O4@SiO2@APTES NPs, and Fe3O4@SiO2@APTES@Glutaryl-PPa MFNPs conjugates were 12 nm, 40 nm, 44 nm, 50 nm in diameter with size distribution standard deviation of 0 nm, 1.2 nm, 0.5 nm, 0.4 nm, respectively. Fig. 3A shows the TEM image of Fe3O4 dispersed in CHCl3, the mean diameters of Fe3O4 NPs were 12 nm with uniform size and spherical shape. The nanoparticles showed narrow size distribution (Fig. 3E) and no agglomeration in CHCl3. However, pure Fe3O4 magnetic nanoparticles has poor dispersity in water and alcohol system because of the magnetic attraction and nanometer effect. Fig. 3B clearly displays that SiO2 shell has successfully coated on the surface of Fe3O4, and the Fe3O4@SiO2 were obtained with a diameter of about 40 nm, and the nanoparticles are spherical, either. But the size distribution is relatively wide (34–43 nm) (Fig. 3F). The average thickness of silica shell is 14 nm approximately. Compared with Fe3O4 in water and alcohol system, Fe3O4@SiO2 NPs shown in Fig. 3B has good dispersity and morphology due to the abundant hydroxyl groups on the surface of SiO2 shell-layer which also reduce magnetic attraction from each other effectively. The TEM image of Fe3O4@SiO2@APTES as shown in Fig. 3C. The thickness of silica shell is markedly thickened and reaches ∼16 nm, because of the hydrophilic amino on the surface of APTES shell-layer and the further reduced magnetic attraction, the Fe3O4@SiO2@APTES NPs are quite homogeneous and exhibit good mono-dispersity with a mean diameter of about 44 nm. These results further proved that APTES has been coated on the Fe3O4@SiO2 surface. Fig. 3D shows the TEM image of Fe3O4@SiO2@APTES@Glutaryl-PPa MFNPs. The thickness of silica shell-layer is markedly thickened and reaches ∼19 nm approximately. After immobilizing PPa of chlorin derivative on the surface of Fe3O4@SiO2@APTES NPs, similar inerratic shape were observed for other magnetic materials and the mean diameters of the MFNPs increased to 50 nm. Because of hydrophobicity of chlorin photosensitizers, the dispersity of the MFNPs reduces slightly, yet the dispersity is good still. The results suggested that PPa has been coated on the surface of silica shell successfully through a bridging glutaryl group.
 |
| | Fig. 3 TEM image and the size distribution of magnetic nanoparticles: Fe3O4 (A, E), Fe3O4@SiO2 (B, F), Fe3O4@SiO2@APTES (C, G), and Fe3O4@SiO2@APTES@Glutaryl-PPa (D, H), respectively. | |
FT-IR spectrum analysis
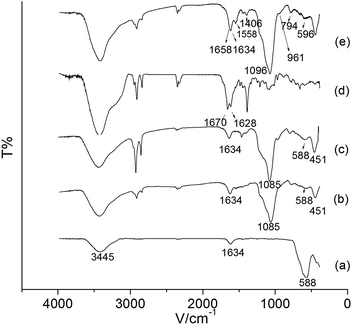
FT-IR was used to monitor the immobilization process. The FT-IR spectrum of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@APTES, PPa and Fe3O4@SiO2@APTES@Glutaryl-PPa NPs are shown in Fig. 4a–e. In Fig. 4a–c, the characteristic absorption band at 588 cm−1 is assigned to the bending vibration of Fe–O in Fe3O4. In Fig. 4b and c, the presence of strong and broad absorption bands at 1085 cm−1 and 451 cm−1 are attributed to the asymmetric stretching and bending vibration of Si–O–Si, respectively. In Fig. 4a–c, the presence of hydroxyl on the surface of NPs and the adsorbed water in the sample are observed in the broad absorption of O–H stretching at 3400–3500 cm−1. Of the characteristic absorption peaks of the primary amine (–NH2), one overlaps with the O–H band at 3400–3500 cm−1, and the other bending vibration of N–H is visible at 1470 cm−1. Also, the peaks at 1400 cm−1 and 1634 cm−1 are attributed to the stretching vibration of C–N and the bending vibration of O–H, respectively. Besides, compared to Fe3O4@SiO2 (Fig. 4b) nanoparticles, the enhanced band between 2950 cm−1 and 2850 cm−1 (Fig. 4c) is attributed to the stretching vibration of the C–H bond in the side chains derived from APTES, which demonstrates the SiO2 layer and APTES layer are coated on the surface of Fe3O4 successfully. Fig. 4d shows the FT-IR spectrum of pure PPa, the band in 1670 cm−1 is attributed to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond. After grafting PPa on the surface of Fe3O4@SiO2@APTES nanoparticles, the absorption peaks of Si–O–Si (asymmetric stretching) and Fe–O (bending vibration) in Fig. 4e move towards high frequency region, appearing at 1096 cm−1 and 596 cm−1, respectively. Also, the absorption peaks at 1658 cm−1, 1558 cm−1 and 1406 cm−1 are in correspondence with vibrations of CO, CC and CN stretching in Fe3O4@SiO2@APTES@Glutaryl-PPa (Fig. 4e), respectively. These peaks confirm the presence of the subunits of chlorin in MFNPs. Overall, these results have provided supportive evidence that PPa had been coated on the surface of Fe3O4@SiO2@APTES successfully.
O bond. After grafting PPa on the surface of Fe3O4@SiO2@APTES nanoparticles, the absorption peaks of Si–O–Si (asymmetric stretching) and Fe–O (bending vibration) in Fig. 4e move towards high frequency region, appearing at 1096 cm−1 and 596 cm−1, respectively. Also, the absorption peaks at 1658 cm−1, 1558 cm−1 and 1406 cm−1 are in correspondence with vibrations of CO, CC and CN stretching in Fe3O4@SiO2@APTES@Glutaryl-PPa (Fig. 4e), respectively. These peaks confirm the presence of the subunits of chlorin in MFNPs. Overall, these results have provided supportive evidence that PPa had been coated on the surface of Fe3O4@SiO2@APTES successfully.
 |
| | Fig. 4 FT-IR spectra of Fe3O4 (a), Fe3O4@SiO2 (b), Fe3O4@SiO2@APTES (c), PPa (d) and Fe3O4@SiO2@APTES@Glutaryl-PPa (e), respectively. | |
Zeta potential measurements
Electrostatic interaction of nanoparticles can be controlled by variation in their surface charges. Therefore, zeta potential measurements of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@APTES and Fe3O4@SiO2@APTES@Glutaryl-PPa (MFNPs) were further used to confirm the presence of amino groups and PPa on the surface of Fe3O4@SiO2 NPs. From Fig. 5, Fe3O4 and Fe3O4@SiO2 show a zeta potential of −18.92 ± 1.54 mV, −9.81 ± 1.29 mV derived from the negative charge of surface of oleic acid and Si–OH, respectively. Meanwhile, the zeta potential of Fe3O4@SiO2@APTES possessed higher positive charge due to the decrease of Si–OH substituted by SiO2–NH2, which increased to 18.68 ± 2.6 mV. After coating PPa on the surface of Fe3O4@SiO2@APTES NPs, the zeta potential decreased to −25.38 ± 2.2 mV as expected, which may be responsible for the prepared MFNPs' highly water dispersity and solubility. In the coupling reaction, the carboxyl group of PPa neutralized the positive charge of the amino group. Based on the above investigation, it is clear that APTES and PPa have been successfully anchored on the surface of Fe3O4@SiO2 NPs.
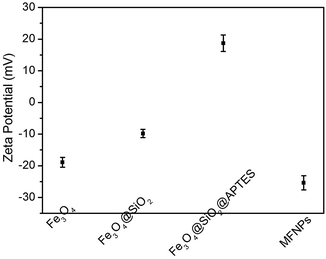 |
| | Fig. 5 Zeta potential of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@APTES, and Fe3O4@SiO2@APTES@Glutaryl-PPa (MFNPs) at pH = 7.0. | |
Magnetic measurements
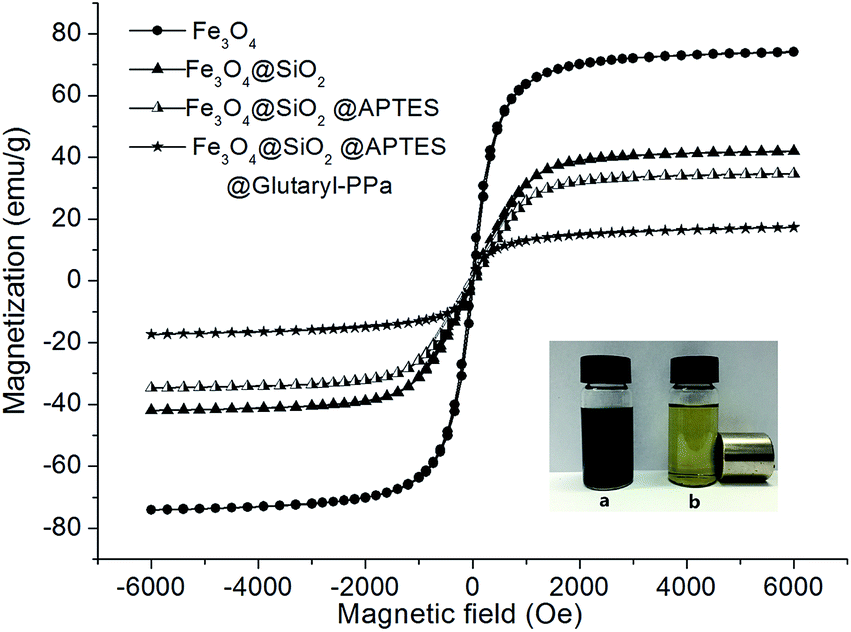
To study the nanoparticles magnetization further, vibrating sample magnetometer (VSM) was performed to investigate the magnetic properties of as-synthesized nanoparticles. Fig. 6 shows the magnetization curves of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@APTES and Fe3O4@SiO2@APTES@Glutaryl-PPa (MFNPs) at 300 K. It can be seen that these nanoparticles have similar super-paramagnetic without magnetic hysteresis phenomenon, and remanent magnetization and coercivity are equal to zero. As shown in Fig. 6, the saturation magnetization of Fe3O4 is about 74.04 emu g−1 at 300 K. After modification of magnetic Fe3O4 surface by SiO2 and APTES, the saturation magnetization of Fe3O4@SiO2 and Fe3O4@SiO2@APTES decreased to 41.92 emu g−1 and 34.65 emu g−1, respectively. While, the saturation magnetization of MFNPs is about 17.31 emu g−1, which is far less than Fe3O4 NPs due to the two coating silica layer and PPa layer.
 |
| | Fig. 6 Magnetization curves at 300 K for Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@APTES and Fe3O4@SiO2@APTES@Glutaryl-PPa MFNPs, respectively. Together with the localization of MFNPs near the magnet (NdFeB). (a) Without magnet; (b) with magnet. | |
The dispersions of the Fe3O4@SiO2@APTES@Glutaryl-PPa are given in Fig. 6a, which was uniformly dispersed in water. The solution was blackish green due to the existence of PPa fragments. The MFNPs nanoparticles rapidly gathered on the side wall of the cylinder under the externally applied magnetic field, as shown in Fig. 6b. The solution became clear and transparent but still exhibited light green. These results indicated that MFNPs have good dispersity and magnetic response in water.
Optical properties of MFNPs
Fig. 7a shows the absorption spectra of PPa and MFNPs. Their visible absorption spectra show an intense Soret band and a shoulder Qy band at 667 nm. The Soret band of PPa and MFNPs are at 408 nm and 405 nm, respectively. Although the absorption spectra of PPa and MFNPs are similar, yet due to the influence of the Fe3O4 NPs, the slight blue shift of the maximum absorption wavelength and the obvious increasement of the baseline in peak pattern could be observed, indicating the presence of PPa on the magnetite nanoparticles. Fig. 7b shows the fluorescence emission spectra of PPa and MFNPs, respectively. The obtained fluorescence spectra of PPa and MFNPs are both symmetric after the excitation at 420 nm, and the maximum emission wavelengths are 676 nm and 672 nm, respectively. The slightly blue shift may be indicating the presence of coupled π–π aggregates at the surface of MFNPs. Moreover, the fluorescence intensity of MFNPs is lower than the fluorescence intensity of PPa, which is due to the reduction of total fluorescent substance content in composite MFNPs nanoparticles under the same concentration.
 |
| | Fig. 7 Ultraviolet-visible absorption spectra (a) and fluorescence mission spectra (b) of PPa and Fe3O4@SiO2@APTES@Glutaryl-PPa, respectively (inset: fluorescence image of MFNPs). The excitation wavelength is 365 nm. | |
The inset shows the fluorescence image of MFNPs dispersed in water, under 365 nm UV irradiation, a tense bright-red fluorescence was observed, which further demonstrates the existence of PPa on the surface of the MFNPs nanoparticles, and shows that the MFNPs has the property of photoluminescence as well.
The spectrum of absorption and fluorescence prove that the presence of superparamagnetic nanoparticles hardly affects the optical properties of chlorin moieties positioned onto the surface of magnetic composites. Only the peak pattern is little altered. In addition, the maximum emission wavelength of MFNPs at 672 nm is sited the range of 650–690 nm, which is considered to be the useful therapeutic region in PDT due to the deep tissue penetration of light.34 Therefore, the spectroscopic property of MFNPs can be exploited to fabricate nanocarriers for photodynamic therapy or medical fluorescence imaging agent.
Differential thermal analysis of MFNPs
Thermal analysis of Fe3O4@SiO2@APTES@Glutaryl-PPa MFNPs was performed to confirm coating formation on the surface of Fe3O4@SiO2. As shown in Fig. 8, with the increase of temperature, the weight of MFNPs gradually decreased. The rate of weight is relatively slow owing to the loss of adsorbed water relatively slow owing to the loss of adsorbed water and residual water below 200 °C. The first weight loss rate is 1.5%. We can see that there are two weight loss steps in the temperature range of 200 °C to 800 °C. The mass loss of about 20% in the range of 200–485 °C owing to the thermal decomposition of PPa and glutaryl, and the maximum weight loss is 485 °C. The second weight loss step in the range of 485–800 °C, the weight loss rate is 3.5%, which is attributed to the decomposition of the residual carbon chain and amide bond. TGA curve shows that PPa was successfully connected to the surface of magnetic Fe3O4@SiO2@APTES by using glutarly as a bridging group, and the total clad ratio approximately reached 20%.
 |
| | Fig. 8 Thermogravimetric analysis (TGA) of Fe3O4@SiO2@APTES@Glutaryl-PPa was performed in the range of 0–800 °C. | |
In vitro photodynamic therapy of MFNPs
Cell uptaking. Before being used as a potential anticancer agent, this photosensitizer-conjugated MFNPs should be able to enter cells. To test if MFNPs meets this prerequisite, cell uptaking experiments were performed on HeLa cells. That is, cells were incubated on 6 well plates (2 × 105 cells per well), followed by incubation overnight. Then added MFNPs (1 mL, 60 μg mL−1) to the cells, incubated for 0.5, 1 and 3 h, respectively. And subsequently analysed by fluorescence imaging. As shown in Fig. 9, the nucleus were stained with DAPI, showing blue fluorescence. After incubated with MFNPs (1 mL, 60 μg mL−1), and being excited by red light (620–760 nm), the cells emitted red fluorescence, which was due to PPa signal of MFNPs. After the DAPI-stained cells were cultured with MFNPs for 30 min at 37 °C, there was no obvious change in the nucleus, but the cells displayed an intense homogeneous cytoplasmic red fluorescence around nucleus, a majority of nucleus were disguised by the red fluorescent cytoplasm, showing pale fuchsia, indicating that MFNPs had penetrated most of cell membrane and entered into the cell. After incubated for 1 h, the nucleus still showed blue fluorescence without any change, while the red fluorescence can be observed from almost all cells, which were nearly identical to the phenomenon of incubation for 3 h, suggesting that MFNPs could permeate the tumour cells quickly, this provided further evidence that MFNPs has suitable lipo-hydro partition coefficient. These results also agree with the ruptured out membrane of cells in the bright field. A recently reported that the second generation photosensitizer chlorin e6 (Ce6) conjugated Fe3O4 is suitable for simultaneous targeting PDT and in vivo MRI.4 Ce6–Fe3O4 NPs may form some small aggregates that can then be internalized by cells and transferred by vesicles before they enter into cells. Also, some nanoparticles enter lysosomes while others array in a line. Because PPa has the same mother nucleus structure of tetraaza[18]annulene with chlorin e6, so we infer that the mechanism of MFNPs cell uptaking may be similar to Fe3O4–Ce6. That is, the nanocarriers or their aggregates are absorbed on the membrane due to their small size effect, and incorporated by endocytosis vesicles through the deformation of the membrane, and subsequently dispersed in cytoplasm of HeLa cells.42 In summary, the magneto-fluorescence material Fe3O4@SiO2@APTES@Glutaryl-PPa MFNPs could enter into the cells quickly and possess suitable lipo-hydro partition coefficient. In addition, after being uptaken, the MFNPs showed marked light red fluorescence in cancer cells, which demonstrated that it’s a potential agent for medical fluorescence imaging.
 |
| | Fig. 9 Fluorescence inverted microscopic images of HeLa cells after incubation with MFNPs (1 mL, 60 μg mL−1) at 37 °C for (A) 30 min, (B) 1 h, (C) 3 h, respectively. The nucleus was stained with DAPI (blue), the red fluorescence was due to PPa signal of MFNPs. The corresponding cells state were observed in bright field. | |
Morphological changes of HeLa cells after PDT
Morphological variation of cells caused by MFNPs-PDT treatment was observed with AO/EB double fluorescent staining. In short, HeLa cells were incubated with MFNPs for 6 h after irradiation for 10 min, and subsequently added AO/EB (5 μL, 100 μg mL−1) dye solution to observe morphological variation. AO dye solution can enter into normal cell and bind to DNA in the nucleus, showing yellow-green fluorescence signal. The reddish orange fluorescence signal was due to the EB dye, which can only enter into the nucleus of necrotic cell. As shown in Fig. 10A, without PDT treatment, the yellow-green fluorescence signal was due to AO bound to DNA of normal cells nucleus, and could be observed from almost all cells. More importantly, cells in Fig. 10A maintained normal appearance, after cultured for additional 6 h, yet reddish orange fluorescence signals were found in most of the cell nucleus in Fig. 10B, and a few appeared yellow-green. It is clear that the nucleus of early apoptotic cells showed yellow-green fluorescence and karyopyknosis could be observed. Meanwhile, as shown in Fig. 10B, the volume increase of necrosis cell and late apoptosis cell could be observed after cultured for 6 h, showing reddish orange fluorescence with unapparent outline. They are becoming dissolved or near disintegration. These combined data clearly and qualitatively indicated that MFNPs-PDT resulted in damage and apoptotic cell death in HeLa cells.
 |
| | Fig. 10 MFNPs-PDT induced damage and apoptotic cell death. HeLa cells were treated without (A) and with (B) MFNPs (1 mL, 50 μg mL−1) with light irradiation for 10 min, and subsequently incubated at 37 °C for 6 h. Morphological variation of cells were studied using AO/EB as nucleus NDA marker, monitored by fluorescence inverted microscopic. | |
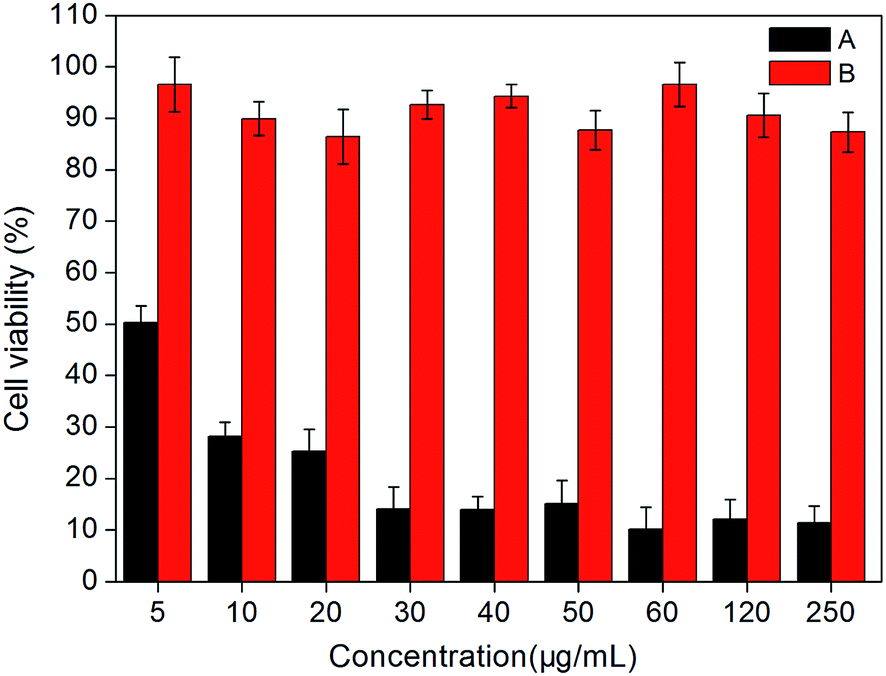
Photodynamic activities. Photodynamic therapy is a method to cure cancer specifically through combining of light and photosensitizer. Photosensitizer with physiological activity is a prerequisite for application in photodynamic therapy, which can be determined by measuring phototoxicity of these particles. The PDT test in this paper was divided into two groups: experimental groups, with cells treated with different concentrations of MFNPs and exposed to light; dark control group keeps identical to the experimental group without irradiation. PDT effect of MFNPs to HeLa cells was detected by MTT assay.Fig. 11 showed the photodynamic activity of the title MFNPs against HeLa cells, the light-dependent cytotoxicity on the cancer cells was significantly different from non-irradiation dark control in statistics, as showed in Fig. 11. With the increase of MFNPs dose, the production of ROS gradually increased, leading to the cell viability gradually decreased. When the concentration of MFNPs reached to 60 μg mL−1, the cells were almost death, the cell viability was reduced to 10.18% and remained stable afterwards, which may be due to that the ROS production increased with increasing MFNPs dose, and reached saturation at a high concentration over a period of time. It is likely that the high concentration of MFNPs are prone to accumulate which may cause the reduction of reactive oxygen species. In addition, 10 μg mL−1 of MFNPs caused approximately 75% cell mortality, suggesting that MFNPs has an obvious photodynamic activity. The dark control group with MFNPs without irradiation revealed that the cell viability was more than 88%, which further suggested that MFNPs had very low toxicity and minimal adverse effects on cancer cells without irradiation. These results demonstrated that MFNPs caused effective cell death with fewer side effects.
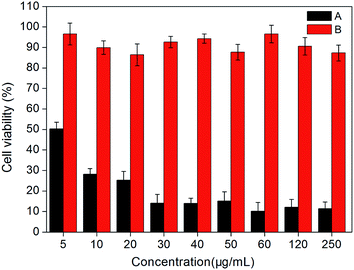 |
| | Fig. 11 Phototoxicity of the title MFNPs against HeLa cells at different concentrations: (A) MTT viability assays with irradiation for 10 min (B) without irradiation. | |
Quenching of active oxygen on PDT (Type I and II reactions). In order to visualize the photochemical processes (Type I and Type II) mechanism of PDT, corresponding ROS of Type I and Type II generated from photodynamic reaction were quenched by using specific quenching agent SA and DM, respectively. The experiments of PDT mechanism research of PDT were divided into four groups: (1) blank control group; (2) MFNPs-PDT group with different concentrations of MFNPs that exposed to light; (3) MFNPs-PDT-SA group in which MFNPs were treated with SA and exposed to light; (4) MFNPs-PDT-DM group in which MFNPs were treated with DM and exposed to light. Precisely, Hela cells were seeded into 96-well plates, and incubated with different concentrations of MFNPs and SA or DM for 4 h after 10 min of irradiation. Fig. 12 showed the influences of three different PDT processing methods on cytotoxicity effects. The cell viability of MFNPs-PDT-SA group and MFNPs-PDT-DM group were obviously higher than that of MFNPs-PDT group, suggesting that the key ROS have been quenched. Furthermore, quenching of singlet oxygen (1O2) generated by Type II photodynamic reaction and oxygen free radicals (hydroxyl radicals, HR) generated by Type I photodynamic reaction can affect apoptotic cell death induced by MFNPs-PDT. In other words, reactive oxygen species have been generated in HeLa cells after treatment with MFNPs-PDT, and the formation may be through two ways, that is, Type I and Type II photodynamic reactions occurred simultaneously. In addition, MFNPs-PDT-SA group was slightly higher than that of MFNPs-PDT-DM group at low concentrations (5–20 μg mL−1) of MFNPs, while at higher concentrations the result was on the contrary. We inferred that Type I reaction plays a predominant role at low concentration of MFNPs, whereas Type II reaction was evoked at higher concentration. Generally, the characteristic of Type I photodynamic reaction is that photosensitizer could interact with any active substrate, producing oxygen free radicals such as hydroxyl radicals (HR), superoxide anion, hydrogen peroxide by electron transfer process in the complex system of MFNPs photodynamic reaction, where there is enough matter can provide some integral substrate for photosensitizer to complete Type I reaction. For Type II reaction, the production of singlet oxygen only requires the presence of tissue oxygen. Therefore, it is reasonable that Type I and Type II reactions occurred simultaneously. In some other cases, participation of both Type I and Type II mechanisms have been invoked and their relative contributions depend on the photosensitizer.43
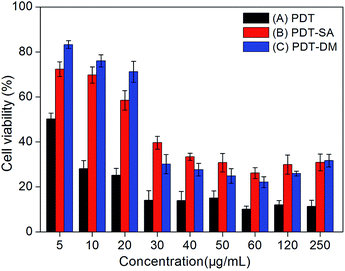 |
| | Fig. 12 The influences of MFNPs-PDT (A), MFNPs-PDT-SA (B), MFNPs-PDT-DM (C) after PDT treatment 24 h on cytotoxicity effects of PDT, respectively. Cell viability was determined by MTT assay. | |
Conclusions
In summary, novel pyropheophorbide-a-conjugated multi-functional magneto-fluorescence nanoparticles Fe3O4@SiO2@APTES@Glutaryl-PPa (MFNPs) were designed and developed successfully. The MFNPs have good water-dispersity, biocompatibility, and strong superparamagnetic, good photoluminescence property. In vitro PDT study suggested that MFNPs mediated PDT significantly inhibited the growth of HeLa cells and displayed high photodynamic therapy activity (cell mortality reached to 89.82%). The cell uptaking tests revealed that MFNPs could enter the cancer cell rapidly, showing that MFNPs have suitable lipo-hydro partition coefficient. We also demonstrated that the formation of reactive oxygen species in HeLa cells after treatment with MFNPs-PDT may be through two mechanisms. That is, Type I photodynamic reactions occurred simultaneously with Type II photodynamic reactions. Our results indicated that MFNPs has great potential application for simultaneous targeting PDT, medical fluorescence imaging, biological probe and MRI.
Acknowledgements
Financial support of this research was provided by National Natural Science Foundation of China (No. 20972036, 21272048), the Natural Science Foundation of Heilongjiang Province (No. B20913) and the Program for Scientific Technological Innovation Team Construction in Universities of Heilongjiang Province (No. 211TD010).
References
- Y. Chen, G. Li and R. K. Pandey, Curr. Org. Chem., 2004, 8, 1105 CrossRef CAS.
- C. A. Robertson, D. Hawkins Evans and H. Abrahamse, J. Photochem. Photobiol., B, 2009, 96, 1 CrossRef CAS PubMed.
- C. H. Sibata, V. C. Colussi, N. L. Oleinick and T. L. Kinsella, Braz. J. Med. Biol. Res., 2000, 33, 869 CAS.
- P. Huang, Z. Li, J. Lin, D. Yang, G. Gao, C. Xu, L. Bao, C. Zhang, K. Wang, H. Song, H. Hu and D. Cui, Biomaterials, 2011, 32, 3447 CrossRef CAS PubMed.
- T. R. Nathan, D. E. Whitelaw, S. C. Chang, W. R. Lees, P. M. Ripley, H. Payne, L. Jones, M. C. Parkinson, M. Emberton, A. R. Gillams, A. R. Mundy and S. G. Bown, J. Urol., 2002, 168, 1427 CrossRef CAS PubMed.
- T.-G. Ahn, B.-R. Lee, E.-Y. Choi, D. W. Kim and S.-J. Han, J. Gynecol. Oncol., 2012, 23, 115 CrossRef PubMed.
- D. E. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380 CrossRef CAS PubMed.
- S. B. Brown, E. A. Brown and I. Walker, Lancet Oncol., 2004, 5, 497 CrossRef CAS PubMed.
- D. Zhang, M. Wu, Y. Y. Zeng, L. J. Wu, Q. T. Wang, X. Han, X. L. Liu and J. F. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 8176 CAS.
- E. S. Nyman and P. H. Hynninen, J. Photochem. Photobiol., B, 2004, 73, 1 CrossRef CAS.
- I. Eichwurzel, H. Stielb and B. Roder, J. Photochem. Photobiol., B, 2000, 54, 194 CrossRef CAS.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995 CrossRef CAS PubMed.
- M. Li, H. Gu and C. Zhang, Nanoscale Res. Lett., 2012, 7, 1 CrossRef PubMed.
- T. K. Jain, J. Richey, M. Strand, D. L. Leslie-Pelecky, C. A. Flask and V. Labhasetwar, Biomaterials, 2008, 29, 4012 CrossRef CAS PubMed.
- G. Cheng, J. L. Zhang, Y. L. Liu, D. H. Sun and J. Z. Ni, Chem. Commun., 2011, 47, 5732 RSC.
- L. Y. Zeng, W. Z. Ren, J. J. Zheng, P. Cui and A. G. Wu, Phys. Chem. Chem. Phys., 2012, 14, 2631 RSC.
- L. Zhang, L. Y. Zeng, Y. W. Pan, S. Luo, W. Z. Ren, A. Gong, X. H. Ma, H. Z. Liang, G. M. Lu and A. G. Wu, Biomaterials, 2015, 44, 82 CrossRef CAS PubMed.
- M. M. Miller, G. A. Prinz, S. F. Cheng and S. Bounoak, Appl. Phys. Lett., 2002, 81, 2211 CrossRef CAS.
- S. Mornet, S. Vasseur, F. Grasset and E. Duguet, J. Mater. Chem., 2004, 14, 2161 RSC.
- T. Charinpanitkul, D. W. Park and K. S. Kim, J. Nanosci. Nanotechnol., 2014, 14, 7995 CrossRef.
- E. Mohsen, J. Jaber, M. A. Mehdi and N. D. Fatemeh, J. Iran. Chem. Soc., 2014, 11, 499 CrossRef CAS.
- C. S. Kumar and F. Mohammad, Adv. Drug Delivery Rev., 2011, 63, 789 CrossRef CAS PubMed.
- B. Samanta, H. H. Yan, N. O. Fischer, S. Jing, J. Joseph and V. M. Rotello, J. Mater. Chem., 2008, 18, 1204 RSC.
- J. Chomoucka, J. Drbohlavova, D. Huska, V. Adam, R. Kizek and J. Hubalek, Pharmacol. Res., 2010, 62, 144 CrossRef CAS PubMed.
- Y. Cheng, R. Q. Yan, W. Y. Wang, Y. Q. Guo, P. Cui and W. J. Song, J. Mater. Sci., 2010, 45, 5347 CrossRef CAS.
- S. J. Zhang, X. H. Liu, L. P. Zhou and W. J. Peng, Mater. Lett., 2012, 68, 243 CrossRef CAS.
- Y. H. Deng, D. W. Qi, C. H. Deng, X. M. Zhang and D. Y. Zhao, J. Am. Chem. Soc., 2008, 130, 28 CrossRef CAS PubMed.
- M. Zubair lqbal, X. H. Ma, T. X. Chen, L. Zhang, W. Z. Ren, L. C. Xiang and A. G. Wu, J. Mater. Chem. B, 2015, 3, 5172 RSC.
- H. L. Ding, Y. X. Zhang, S. Wang, J. M. Xu, S. C. Xu and G. H. Li, Chem. Mater., 2012, 24, 4572 CrossRef CAS.
- M. Ghavami, M. Kooki and M. Z. Kassaee, J. Chem. Sci., 2012, 125, 1347 CrossRef.
- M. Perrier, M. Gary-Bobo, L. Lartigue, D. Brevet, A. Morere, M. Garcia, P. Maillard, L. Raehm and Y. Guari, J. Nanopart. Res., 2013, 15, 1602 CrossRef.
- L. Y. Zeng, W. Z. Ren, L. C. Xiang, J. J. Zheng, B. Chen and A. G. Wu, Nanoscale, 2013, 5, 2107 RSC.
- H. W. Gu, K. M. Xu, Z. M. Yang, C. K. Chang and B. Xu, Chem. Commun., 2005, 4270 RSC.
- M. Zubair lqbal, X. H. Ma, T. X. Chen, L. Zhang, W. Z. Ren, L. C. Xiang and A. G. Wu, J. Mater. Chem. B, 2015, 3, 5172 RSC.
- J. N. Park, K. J. An, Y. S. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, N.-M. Hwang and T. H. Hyeon, Nat. Mater., 2004, 3, 891 CrossRef CAS PubMed.
- Z. T. Liu, L. Xiong, Z. P. Liu, X. Y. Miao, L. W. Lin and Y. Wen, Nanoscale Res. Lett., 2014, 9, 319 CrossRef PubMed.
- E. K. Lim, E. Jang, B. Kim, J. Choi, K. Lee, J. S. Suh, Y. M. M. Huh and S. Haam, J. Mater. Chem., 2011, 21, 12473 RSC.
- Y. Hyunhee, M. Seung-Kwan, H. Taewon, K. Y. Seok, K. Joo-Hwan, C. Sung-Wook and K. J. Hyun, Langmuir, 2013, 29, 5962 CrossRef PubMed.
- S. F. Mujtaba, A. Dwivedi, N. Yadav, R. S. Ray and G. Singh, J. Hazard. Mater., 2013, 252–253, 258 CrossRef CAS PubMed.
- D. Y. Yu, G. H. Huang, F. X. Xu, B. S. Ge, S. Liu, H. Xu and F. Huang, Photosynth. Res., 2014, 122, 203 CrossRef CAS PubMed.
- D. B. Jennings, M. E. Ehrenshaft, D. M. Pharr and J. D. Williumson, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 15129 CrossRef CAS.
- S. Zhang, X. J. Chen, C. R. Gu, Y. Zhang, J. D. Xu, Z. P. Bian, D. Yang and N. Gu, Nanoscale Res. Lett., 2009, 4, 70 CrossRef CAS.
- H. Liyi, Y. Yi, K. Yuichiro, Z. Timur, T. Masamitsu and H. R. Michael, Lasers Surg. Med., 2012, 44, 490 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.