
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the high-temperature properties of Pr2NiO4+δ by simultaneous Pr- and Ni-cation replacement†
S. Ya.
Istomin
 *a,
O. M.
Karakulina
a,
M. G.
Rozova
a,
S. M.
Kazakov
a,
A. A.
Gippius
ab,
E. V.
Antipov
a,
I. A.
Bobrikov
c,
A. M.
Balagurov
c,
A. A.
Tsirlin
de,
A.
Michau
f,
J. J.
Biendicho
fgh and
G.
Svensson
f
*a,
O. M.
Karakulina
a,
M. G.
Rozova
a,
S. M.
Kazakov
a,
A. A.
Gippius
ab,
E. V.
Antipov
a,
I. A.
Bobrikov
c,
A. M.
Balagurov
c,
A. A.
Tsirlin
de,
A.
Michau
f,
J. J.
Biendicho
fgh and
G.
Svensson
f
aM.V. Lomonosov Moscow State University, Moscow 119991, Russia. E-mail: istomin@icr.chem.msu.ru
bA.V. Shubnikov Institute of Crystallography, Moscow 117333, Russia
cJoint Institute for Nuclear Research, Dubna 141980, Russia
dNational Institute of Chemical Physics and Biophysics, 12618 Tallinn, Estonia
eExperimental Physics VI, Centre for Electronic Correlations and Magnetism, University of Augsburg, 86159 Augsburg, Germany
fDepartment of Materials and Environmental Chemistry, Stockholm University, S-106 91 Stockholm, Sweden
gThe ISIS Facility, STFC Rutherford Appleton Laboratory, Didcot OX110 QX, Oxfordshire, UK
hIREC, Catalonia Institute for Energy Research, Jardins de les Dondes de Negre, e1, 08930 Sant Adria del Besos, Spain
First published on 30th March 2016
Abstract
Novel Pr2−xSrxNi1−xCoxO4±δ (x = 0.25; 0.5; 0.75) oxides with the tetragonal K2NiF4-type structure have been prepared. Room-temperature neutron powder diffraction (NPD) study of x = 0.25 and 0.75 phases together with iodometric titration results have shown the formation of hyperstoichiometric oxide for x = 0.25 (δ = 0.09(2)) and a stoichiometric one for x = 0.75. High-temperature X-ray powder diffraction (HT XRPD) showed substantial anisotropy of the thermal expansion coefficient (TEC) along the a- and c-axis of the crystal structure, which increases with increasing the Co content from TEC(c)/TEC(a) = 2.4 (x = 0.25) to 4.3 (x = 0.75). High-temperature NPD (HT NPD) study of the x = 0.75 sample reveals that a very high expansion of the axial (Ni/Co)–O bonds (75.7 ppm K−1 in comparison with 9.1 ppm K−1 for equatorial ones) is responsible for such behaviour, and is caused by a temperature-induced transition between low- and high-spin states of Co3+. This scenario has been confirmed by high-temperature magnetization measurements on a series of Pr2−xSrxNi1−xCoxO4±δ samples. For compositions with high Ni content (x = 0.25 and 0.5) we synthesised K2NiF4-type oxides Pr2−x−ySrx+y(Ni1−xCox)O4±δ, y = 0.0–0.75 (x = 0.25); y = 0.0–0.5 (x = 0.5). The studies of the TEC, high-temperature electrical conductivity in air, chemical stability of the prepared compounds in oxygen and toward interaction with Ce2−xGdxO2−x/2 (GDC) at high temperatures reveal optimal behaviour of Pr1.35Sr0.65Ni0.75Co0.25O4+δ. This compound shows stability in oxygen at 900 °C and does not react with GDC at least up to 1200 °C. It features low TEC of 13 ppm K−1 and high-temperature electrical conductivity in air of 280 S cm−1 at 900 °C, thus representing a promising composition for use as a cathode material in intermediate temperature solid oxide fuel cells (IT-SOFC).
Introduction
Layered nickelates R2NiO4+δ (R = La, Pr, Nd) have been considered as promising cathode materials for intermediate temperature (750–900 °C) solid oxide fuel cells (IT-SOFC) due to their high electrical conductivity, fast oxygen diffusion and thermal expansion coefficient (TEC) compatible with typical SOFC electrolytes.1 The crystal structure of these nickelates belongs to the K2NiF4 type and consists of alternate rock-salt and perovskite slabs (Fig. 1). Due to a size mismatch between metal–oxygen distances of Ni2+ and rare earth cations, the NiO6 octahedra tilt, which results in a lowering of the crystal structure symmetry from tetragonal (ideal K2NiF4-type structure) to orthorhombic.2,3 Incorporation of extra oxygen atoms (δ), occupying the tetrahedral voids in the rock-salt slab (Fig. 1) reduces this mismatch due to a decrease of the nickel ionic radius caused by partial oxidation of Ni2+ to Ni3+.4–6 The amount of hyperstoichiometric oxygen δ depends on the rare-earth cation (R). The smaller the radius of the R cations, the higher the oxygen content (R = La, δ = 0.16;7 R = Pr, Nd, δ = 0.21–0.23 (ref. 5, 6 and 8)) for phases synthesized at the same conditions. The hyperstoichiometric oxygen atoms can diffuse by an interstitial mechanism within the rock-salt slab of the K2NiF4-type structure involving as well apical oxygen atoms.9 This results in higher oxygen diffusion in rare-earth nickelates R2NiO4+δ (R = La, Pr, Nd) in comparison with traditional IT-SOFC cathode material like (La,Sr)(Co,Fe)O3−δ (LSFC), with the perovskite structure. The highest oxygen diffusion (D* = 2.5 × 10−8 cm2 s−1) and surface exchange (k = 5 × 10−7 cm s−1) coefficients at 600 °C have been observed in Pr2NiO4+δ.1 Other advantages of Pr2NiO4+δ are the highest electrical conductivity among other rare-earth nickelates and low TEC. However, it is unstable in oxygen in comparison with La2NiO4+δ and Nd2NiO4+δ and decomposes with the formation of Pr4Ni3O10 and Pr6O11 at 850 °C.10 | ||
| Fig. 1 The crystal structure of (Pr,Sr)2(Ni,Co)O4+δ (K2NiF4-type). Hyperstoichiometric oxygen atoms (δ) are located in the tetrahedral vacancies. | ||
Both crystal structure and high-temperature properties of R2NiO4 oxides can be modified by cation substitution in the A- and B-sublattices. Partial replacement of rare-earth cations by larger Sr2+ according to the chemical formula R2−xSrxNiO4+δ resulted in the formation of solid solutions with tetragonal K2NiF4 structure. A solid solution with tetragonal symmetry is observed for x = 0.2–1.0 for R = La,11 Pr,7 Nd,12 and from 0.8 to 1.0 for Sm.7 The increase of Sr content is accompanied by a decrease of the hyperstoichiometric oxygen content giving rise to oxygen-deficient phases. The range of x in which δ is negative is not well defined and varies depending upon the synthesis conditions.8,13,14 The TEC drastically increases with increasing of Sr-content up to 16 ppm K−1 for LaSrNiO4+δ.15 Meanwhile, high-temperature electrical conductivity increases reaching 273 S cm−1 at 600 °C for x = 0.75. A similar trend is observed for Pr2−xSrxNiO4+δ, where the conductivity is higher than 100 S cm−1 for the substituted phases (x ≥ 0.5).16,17 The oxygen diffusion coefficient D* is the highest for unsubstituted La2NiO4+δ and decreases with increasing the Sr-content.18,19 However, recently an increase of D* has been observed for compositions with high amount of Sr (x = 0.8).19
With the aim to improve the chemical stability in oxygen, high-temperature electrical conductivity and electrocatalytic activity of R2NiO4+δ for oxygen reduction in the present work we introduced Co3+ cations in Pr2NiO4+δ. This was performed by the simultaneous replacement of the equal amounts of Pr and Ni cations by Sr and Co, respectively, according to the chemical formula Pr2−xSrxNi1−xCoxO4±δ (x = 0.25; 0.5; 0.75; 1.0). For compositions with high Ni-content (x = 0.25 and 0.5) we synthesised Pr2−x−ySrx+y(Ni1−xCox)O4±δ, y = 0.0–0.75 (x = 0.25); y = 0.0–0.5 (x = 0.5) where the oxidation state of Ni has been varied by partial replacement of Pr3+ by Sr2+. Effects of cations substitutions on the crystal structure, high-temperature electrical conductivity in air and thermal expansion behaviour of the novel compounds were investigated.
Experimental
Pr2−xSrxNi1−xCoxO4±δ (x = 0.25; 0.5; 0.75; 1.0) and Pr2−x−ySrx+y(Ni1−xCox)O4±δ, y = 0.0–0.75 (x = 0.25); y = 0.0–0.5 (x = 0.5) samples were prepared by conventional solid-state route in air at 1300–1350 °C, 20–24 h with preliminary annealing and regrinding at 950 °C, 24 h. Pr6O11, SrCO3, NiO and Co3O4 (all analytical grade) were used as initial reagents. The densities of the ceramic samples were measured by pycnometry.Phase purity of the compounds was checked by X-ray powder diffraction (XRPD) recorded on Huber G670 Guinier diffractometer (CuKα1 radiation, image foil detector). High-temperature X-ray powder diffraction (HT XRPD) data in air were collected by Bruker D8-Advance diffractometer (CuKα1 radiation, LynxEye PSD) in reflection mode equipped with high-temperature camera XRK-900 (Anton Paar). Unit cell parameters were refined by Rietveld method using TOPAS-3 program package. Oxygen content of the single-phase samples was determined by iodometric titration.
The thermal expansion behaviour of the samples was monitored by Netzsch 402C dilatometer in air at 25–900 °C with the heating rate of 10 K min−1. TG studies were performed in artificial air (20% O2(g), 80% Ar(g)) from 25 to 900 °C with a heating rate of 10 K min−1 by Netzsch STA 449C thermoanalyser.
High-temperature electrical conductivity of ceramic samples was measured in air by a standard 4-probe method in the temperature range of 25–900 °C. The samples had the shape of a disk with ∼16–18 mm diameter and 1–2 mm thickness. The contacts were made from platinum wire (d ∼ 0.2 mm) placed in alumina tube and were pressed independently to the surface of the sample by the separate individual springs situated at the top of the quartz sample holder kept at room temperature. The contacts were arranged in a line with 5–6 mm spacing between them. The influence of undesirable thermoelectric power was omitted by subtracting two successive voltage values on the potential contacts (the inner pair) measured at opposite current directions. The resulting resistivity value was recalculated into specific resistance using the approach developed in ref. 20.
Neutron powder diffraction data were collected using the POLARIS diffractometer at the ISIS pulsed spallation neutron source, Rutherford Appleton Laboratory, UK. Approximately 3 g of sample were loaded into a thin-walled cylindrical vanadium can and mounted in an automatic sample changer. Prior to analysis, the data were normalized using an incoherent scattering pattern from a vanadium sample. Structural models were tested by refinement of the neutron diffraction data using GSAS program package.21
High-temperature neutron diffraction (HT NPD) data were collected using high-resolution Fourier powder diffractometer (HRFD)22 at IBR-2 pulsed reactor in Dubna (Russia). Rietveld analysis of the data was performed using MRIA program package.23 Data were collected at room temperature and at 100–500 °C in air with a step 100 °C.
High-temperature magnetic susceptibility measurements were performed on pressed pellets of Pr2−xSrxNi1−xCoxO4±δ (x = 0.0, 0.25; 0.5; 0.75) in the temperature range 10–800 K in applied magnetic fields up to 8 T using Quantum Design PPMS equipped with a vibrating sample magnetometer (VSM) option and oven setup. The measurements above 400 K were performed in high vacuum under constant gas pressure of 10−5 Torr. The samples were stable under these conditions, as confirmed by the excellent reproducibility of the data obtained during heating and cooling sweeps.
Results and discussion
Pr2−xSrxNi1−xCoxO4±δ (x = 0.25; 0.5; 0.75; 1.0)
| x | a (Å) | c (Å) | Oxygen content (4 ± δ) | B-Cation oxidation state |
|---|---|---|---|---|
| 0.25 | 3.8171(2) | 12.3838(8) | 4.11(1) | +2.47(2) |
| 0.50 | 3.8049(1) | 12.3201(6) | 4.05(3) | +2.60(6) |
| 0.75 | 3.7947(1) | 12.3171(5) | 3.99(2) | +2.73(4) |
| 1.0 | 3.7787(2) | 12.3415(7) | 3.97(2) | +2.94(4) |
Crystal structures of the x = 0.25 and 0.75 phases were refined using neutron powder diffraction data collected at room temperature. No reflection splitting was observed in both diffraction patterns, thus confirming the tetragonal crystal structure with S.G. I4/mmm. Reflections from admixtures of CoO and NiO were observed in NPD patterns of both compounds. However, their refined content was too small to have significant influence on the structure refinement. The refined weight fractions of NiO and CoO in the x = 0.25 sample were 0.0090(2) and 0.0035(5), and for x = 0.75–0.0029(2) and 0.0033(6), respectively. Observed, calculated, and difference NPD patterns for the x = 0.25 sample are given on Fig. S1.†
Crystal structure refinement for the x = 0.25 phase shows the presence of additional oxygen atoms positioned in the tetrahedral voids within the rock-salt slab (site O3 at 4d (0, 1/2, 1/4), Fig. 1). The oxygen content of the x = 0.25 phase calculated from the refined occupancy of this oxygen position (Table S1†) corresponds to Pr1.75Sr0.25Ni0.75Co0.25O4.09±0.02 in excellent agreement with the results from the iodometric titration. No additional oxygen atoms were found at the 4d site in the crystal structure of x = 0.75 phase, in accordance with chemical titration results (Table 1). Selected interatomic distances in the crystal structures of x = 0.25 and 0.75 phases are given in Table S2.† Anisotropic atomic displacement parameters (ADP) for all atoms, except for O3, were used for the refinement of the crystal structures. Atomic displacement ellipsoids in the crystal structure of the x = 0.25 phase are shown in Fig. S2.† Substantial anisotropy of the oxygen atoms ADPs was observed in the crystal structure of the x = 0.25 phase. U33 for the equatorial oxygen atom O1 and U11 for the axial oxygen atom O2 are several times higher in comparison with those in the x = 0.75 phase (Table S1†). The most plausible explanation for such behaviour is the influence of the additional oxygen atom in the O3 position of the crystal structure for x = 0.25.
 | ||
| Fig. 2 Thermal expansion curves for Pr2−xSrxNi1−xCoxO4±δ (x = 0.25, 0.50, 0.75, 1.0) measured in air. | ||
Large discrepancy between the values of TEC calculated from dilatometry data and from HT XRPD is observed for studied compounds (Table 2). This is especially the case for compositions with x ≥ 0.5 containing large amounts of Co. The difference between the TEC values determined by dilatometry and HT HRPD approaches ∼60% for the x = 0.75 sample. Such situation is very similar to what was observed and discussed in ref. 25, where the discrepancy was attributed to the large anisotropy of the thermal expansion along the a- and c-axes of the crystal structure. In this case large thermal stress of the ceramic sample is relaxed by the presence of internal micro cracks.
| x | TEC(a) | TEC(c) | TEC(c)/TEC(a) | TEC (V1/3) | TECdil |
|---|---|---|---|---|---|
| 0.25 | 11.3 | 27.3 | 2.4 | 16.5 | 14.1 |
| 0.50 | 9.7 | 40.1 | 4.1 | 19.3 | 13.1 |
| 0.75 | 10.9 | 46.4 | 4.3 | 22.1 | 13.7 |
| 1.0 | 14.1 | 44.9 | 3.2 | 24.2 | 13.3 |
 | ||
| Fig. 3 Temperature dependence of the unit cell parameters for x = 0.75 phase derived from HT NPD data collected using HRFD diffractometer. Unit cell parameters were normalized by the unit cell parameter values at 295 K. Dashed line shows a linear fit of the c-parameter. | ||
The expansion of (Ni/Co)–O2 and (Pr/Sr)–O2 bonds gives the highest contribution to the thermal expansion along the c-axis (Fig. 1). The temperature dependence of the (Ni/Co)–O2 interatomic distances is shown in Fig. 4a. TEC calculated for the expansion of axial (Ni/Co)–O2 bonds in studied temperature range is 75.7 ppm K−1, while it is 9.1 ppm K−1 for the equatorial (Ni/Co)–O1 one. Therefore, a major contribution to the large expansion along the c-axis in the x = 0.75 phase is given by the expansion of the axial (Ni/Co)–O2 bond, whereas nearly no change of the (Pr/Sr)–O2 bond with temperature is observed (Fig. 4b). This situation is very similar to previous reports for La2−xSrxCoO4 (ref. 26) and Pr1.25Sr0.75Cu0.25Co0.75O3.95(2).25 It was proposed that a temperature-induced transition between low- and high-spin states of Co3+ is responsible for such behaviour.
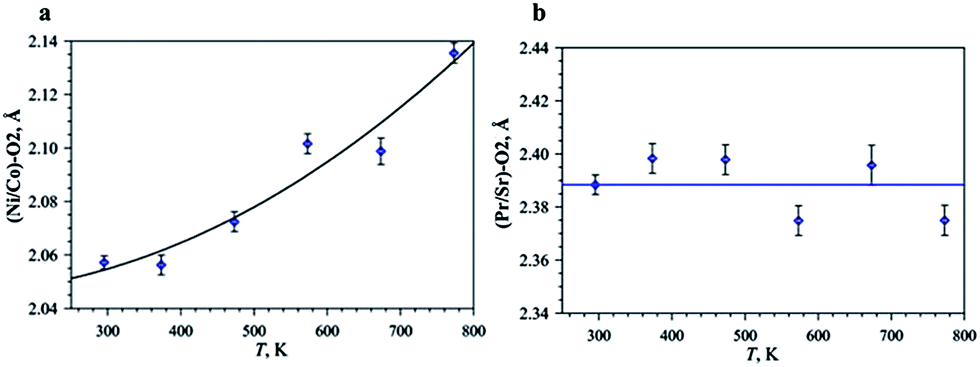 | ||
| Fig. 4 Temperature dependence of the (Ni/Co)–O2 (a) and (Pr/Sr)–O2 (b) interatomic distances. | ||
We were able to confirm this scenario by high-temperature magnetization measurements on a series of Pr2−xSrxNi1−xCoxO4±δ samples (Fig. 5). All of the samples revealed paramagnetic-like behaviour with no appreciable field dependence. The linear part of the inverse susceptibility was fitted by the Curie–Weiss law χ = C/(T + θ), where C = NAμeff2/3kB is the Curie constant yielding the paramagnetic effective moment μeff, and θ is the Curie–Weiss temperature. The fitted parameters are listed in Table 3 along with expected spin-only values, where we assumed Pr3+ (3.58 μB), Ni2+ (2.83 μB), and high-spin Ni3+ (3.87 μB) with the Ni2+/Ni3+ ratio fixed by the oxygen stoichiometry. For Co-doped samples, both low-spin (0 μB) and high-spin (4.90 μB) regimes of Co3+ were considered.
 | ||
| Fig. 5 Inverse magnetic susceptibility (1/χ) of the samples with x = 0.0, 0.25, 0.50, and 0.75 measured in the applied field of 0.5 T. Lines are Curie–Weiss fits to the data yielding the parameters listed in Table 5. | ||
| x | μ eff (experiment), μB | μ eff (calculated), μB | θ, K |
|---|---|---|---|
| 0.00 | 6.24 | 6.06 | 111 |
| 0.25 | 5.23 | 5.47 | 29 |
| 0.50 | 5.08/6.21 | 4.89/5.99 | 26/253 |
| 0.75 | 4.76/6.21 | 4.25/6.00 | 37/382 |
The x = 0 sample revealed an effective moment of 6.24 μB per f.u. that can be understood as a combination of two Pr3+ ions and a mixture of Ni2+/Ni3+ yielding μeff = 6.06 μB. The much lower effective moment of the x = 0.25 sample (μeff = 5.23 μB per f.u.) reflects the substitution of Pr3+ by non-magnetic Sr2+ and of Ni2+/Ni3+ by non-magnetic low-spin Co3+.
The high-temperature behaviour of the x = 0.5 and 0.75 samples is more complex. Here, one can identify two linear regions. Below 300–350 K, 1/χ reveals effective moments, which are even lower than in the x = 0.25 sample and can be accounted by assuming that only Pr3+ and the residual Ni2+ are magnetic. Above 500 K, another linear region is observed, where the effective moment is around 6 μB and the Curie–Weiss temperature is much higher than at low temperatures. Both observations suggest that at high temperatures Co3+ becomes magnetic, thus triggering strong interactions within the octahedral layers, while at low temperatures magnetic interactions measured by the Curie–Weiss temperature θ are on the order of 40–50 K and characteristic of the weak 4f magnetism.
These results show that the anomalously high expansion of the (Ni,Co)–O2 axial bond can be explained by a temperature-induced transition between low- and high-spin states of Co3+ which occurs at T > 500 K for the x = 0.5 and 0.75 phases. High-temperature NPD study shows that the calculated linear TEC of the (Ni,Co)–O2 axial bond for the x = 0.75 phase is 55 ppm K−1 in the temperature range 295–473 K and 87 ppm K−1 at 473–773 K.
Altogether we can conclude that high anisotropy of the thermal expansion of Pr2−xSrxNi1−xCoxO4±δ oxides with high Co-content (x ≥ 0.5) hampers their use in high-temperature electrochemical devices since accumulation of the thermal stresses may results in their crumbling upon thermal cycling. For the phase with low Co-content (x = 0.25) another drawback was revealed. As mentioned above, one of the disadvantages of undoped Pr2NiO4+δ is its instability in oxygen at T > 850 °C, where it decomposes into Pr4Ni3O10 and Pr6O11.10,27,28 The treatment of the x = 0.25 phase in oxygen flow at 900 °C for 24 h revealed the partial oxidation into Pr6O11 and PrNiO3. In order to improve properties of Pr2−xSrxNi1−xCoxO4±δ oxides with low Co-content we prepared solid solutions Pr2−x−ySrx+y(Ni1−xCox)O4±δ, y = 0.0–0.75 (x = 0.25); y = 0.0–0.5 (x = 0.5) where we further increased the average oxidation state of Ni toward +3.
Pr2−x−ySrx+y(Ni1−xCox)O4±δ, y = 0.0–0.75 (x = 0.25); y = 0.0–0.5 (x = 0.5)
 | ||
| Fig. 6 Unit cell parameters a (left) and c (right) versus Sr content Pr2−x−ySrx+y(Ni1−xCox)O4±δ, y = 0.0–0.75 (x = 0.25); y = 0.0–0.5 (x = 0.5). The error values of the unit cell parameters are smaller that the symbols of the data points. | ||
A bell-like dependence of the c-parameter over Sr-content with the maximum at y = 0.4 (Pr1.35Sr0.65Ni0.75Co0.25O4+δ) is clearly observed for the x = 0.25 series, while it is less pronounced for the x = 0.5 series with a maximum at y = 0.3 (Pr1.2Sr0.8Ni0.5Co0.5O4±δ) (Fig. 6). At the same time, the a-parameter shows minimum for these compositions. Such a behaviour was also reported in the literature for the hole-doped nickelates R2−xSrxNiO4±δ, R = La, Pr and Nd and can be explained by the transformation from t62g(z2)1 to t62g(x2 − y2)1 electron configuration of Ni3+.30
For further studies we selected Pr1.75−ySr0.25+yNi0.75Co0.25O4±δ compositions since Ni-rich oxide (y = 0.0) shows lowest TEC value (Table 2). Iodometric titration of the composition with the maximum c-axis (Pr1.35Sr0.65Ni0.75Co0.25O4±δ) showed the formation of a nearly oxygen stoichiometric phase with the oxygen content (4 + δ) = 4.01(2).
For Pr1.35Sr0.65Ni0.75Co0.25O4±δ (y = 0.40) both chemical stability in oxygen atmosphere and toward chemical interaction with standard electrolyte materials for SOFC like GDC and YSZ were studied. In comparison with Pr1.75Sr0.25Ni0.75Co0.25O4±δ phase (y = 0.0), no decomposition was observed after heat treatment in oxygen gas flow at 900 °C for 24 h. It should be noted that both unit cell parameters and oxygen content remained unchanged during such treatment. The higher stability of the y = 0.40 sample in oxygen is most likely related to the higher oxidation state of nickel cations (+2.56 assuming Co3+ and Pr3+) in comparison with +2.29 in the y = 0.0 sample, which prevents further oxidation of nickel.
For the stability test towards chemical interaction with GDC and YSZ, powder of Pr1.35Sr0.65Ni0.75Co0.25O4±δ was mixed with electrolytes in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mass ratios and annealed at 800–1100 °C with a step of 100 °C during 50 h. An XRPD study revealed vigorous reactivity with YSZ, which started already at 900 °C with the formation of Pr2Zr2O7 and SrZrO3 (see Fig. S3a†). However, no admixture phases were observed after heating Pr1.35Sr0.65Ni0.75Co0.25O4±δ/GDC mixture up to 1200 °C (see Fig. S3b†). Therefore, Pr1.35Sr0.65Ni0.75Co0.25O4±δ oxide may be used as cathode either with GDC electrolyte directly or with the YSZ electrolyte separated by the protective layer of GDC.
:1 mass ratios and annealed at 800–1100 °C with a step of 100 °C during 50 h. An XRPD study revealed vigorous reactivity with YSZ, which started already at 900 °C with the formation of Pr2Zr2O7 and SrZrO3 (see Fig. S3a†). However, no admixture phases were observed after heating Pr1.35Sr0.65Ni0.75Co0.25O4±δ/GDC mixture up to 1200 °C (see Fig. S3b†). Therefore, Pr1.35Sr0.65Ni0.75Co0.25O4±δ oxide may be used as cathode either with GDC electrolyte directly or with the YSZ electrolyte separated by the protective layer of GDC.
 | (1) |
 | ||
| Fig. 7 Temperature dependence of electrical conductivity for Pr1.75−ySr0.25+y(Ni0.75Co0.25)O4±δ, y = 0.0, y = 0.15 and 0.40. Red lines show fit of the experimental data according to eqn (1), while black symbols are experimental points. | ||
| y | σ exp, 700 °C | σ exp, 900 °C | σ cor, 700 °C | σ cor, 900 °C | E a, eV | Density, % |
|---|---|---|---|---|---|---|
| 0.0 | 43 | 56 | 61 | 80 | 0.18 | 72 |
| 0.15 | 28 | 30 | 50 | 54 | 0.13 | 56 |
| 0.40 | 260 | 280 | 293 | 315 | 0.15 | 90 |
Owing to the substantially different porosity of the samples (Table 5), we recalculated electrical conductivity values taking the porosity into account using the formula from ref. 33:
 | (2) |
Conclusions
By simultaneous replacement of Ni for Co and Pr for Sr in Pr2NiO4+δ we have successfully prepared Pr2−xSrxNi1−xCoxO4±δ (x = 0.25; 0.5; 0.75) oxides with the tetragonal K2NiF4-type structure. Similar to other Co-containing oxides with the K2NiF4-type structure, Pr2−xSrxNi1−xCoxO4±δ (x = 0.25; 0.5; 0.75) show substantial anisotropy of the thermal expansion along the a- and c-axes of the crystal structure, and this anisotropy increases upon Co substitution. This results in a rather large difference between the TEC values calculated from dilatometry and HT XRPD data. Using a combination of HT NPD and HT magnetization measurements, we showed for the first time that such anisotropy of thermal expansion is most likely associated with the temperature-induced transition between low- and high-spin states of Co3+ cations.In order to further vary the oxidation state of Ni in Pr2−xSrxNi1−xCoxO4±δ, x = 0.25 and 0.5 oxides, we replaced Pr3+ by Sr2+. This allowed to prepare Pr1.35Sr0.65Ni0.75Co0.25O4±δ composition with very promising properties as cathode material in IT-SOFC. This material shows the TEC of 13.0 ppm K−1 comparable with that of GDC electrolyte, as well as the chemical stability in oxygen at 900 °C as well as towards interaction with GDC up to 1250 °C. This composition exhibits high values of high-temperature electrical conductivity in air approaching 300 S cm−1 at 900 °C and is considered as promising cathode material in IT-SOFC.
Acknowledgements
This work was partially supported by Russian Foundation for Basic Research (grants no. 14-03-01083, 14-29-04091 ofi_m), by Skolkovo Institute of Science and Technology (Center of Electrochemical Energy) and MSU-development Program up to 2020. AAT acknowledges partial financial support by the Federal Ministry of Science and Education through the Sofja Kovalevskaya Award of Alexander von Humboldt Foundation.Notes and references
- E. Boehm, J.-M. Bassat, P. Dordor, F. Mauvy, J.-C. Grenier and P. Stevens, Solid State Ionics, 2005, 176, 2717 CrossRef CAS.
- J. D. Sullivan, D. J. Buttrey, D. E. Cox and J. Hriljac, J. Solid State Chem., 1991, 94, 337 CrossRef CAS.
- R. Saez Puche, F. Fernández, J. Rodríguez Carvajal and J. L. Martínez, Solid State Commun., 1989, 72, 273 CrossRef CAS.
- H. Tamura, A. Hayashi and Y. Ueda, Phys. C, 1996, 258, 61 CrossRef CAS.
- Y. Toyosumi, H. Ishikawa and K. Ishikawa, J. Alloys Compd., 2006, 408–412, 1200 CrossRef CAS.
- E. Niwa, K. Wakai, T. Hori, K. Yashiro, J. Mizusaki and T. Hashimoto, Thermochim. Acta, 2014, 575, 129 CrossRef CAS.
- S. C. Chen, K. V. Ramanujachary and M. Greenblatt, J. Solid State Chem., 1993, 105, 444 CrossRef CAS.
- C. Allançon, P. Odier, J. M. Bassat and J. P. Loup, J. Solid State Chem., 1997, 131, 167 CrossRef.
- A. Chroneos, D. Parfitt, J. A. Kilner and R. W. Grimes, J. Mater. Chem., 2010, 20, 266 RSC.
- P. Odier, C. Allanion and J.-M. Bassat, J. Solid State Chem., 2000, 153, 381 CrossRef CAS.
- J. E. Millburn, M. A. Green, D. A. Neumann and M. J. Rosseinsky, J. Solid State Chem., 1999, 145, 401 CrossRef CAS.
- Y. Takeda, M. Nishijima, N. Imanishi, R. Kanno, O. Yamamoto and M. Takano, J. Solid State Chem., 1992, 96, 72 CrossRef CAS.
- Y. Takeda, R. Kanno, M. Sakano, O. Yamamoto, M. Takano, Y. Bando, H. Akinaga, K. Takita and J. B. Goodenough, Mater. Res. Bull., 1990, 25, 293 CrossRef CAS.
- M. J. Sayagues, M. Vallet-Regi, A. Caneiro and J. M. Gonzalez-Calbet, Solid State Ionics, 1993, 66, 21–26 CrossRef CAS.
- A. Aguadero, M. J. Escudero, M. Perez, J. A. Alonso, V. Pomjakushin and L. Daza, Dalton Trans., 2006, 4377 RSC.
- J. Yang, J. Cheng, Q. Jiang, Y. Wang, R. Wang and J. Gao, Int. J. Hydrogen Energy, 2012, 37, 1746 CrossRef CAS.
- V. Vashook, J. Zosel, T.-L. Wen and U. Guth, Solid State Ionics, 2006, 177, 1827 CrossRef CAS.
- S. J. Skinner and J. A. Kilner, Solid State Ionics, 2000, 135, 709 CrossRef CAS.
- T. Inprasit, S. Wongkasemjit, S. J. Skinner, M. Burriel and P. Limthongkul, RSC Adv., 2015, 5, 2486 RSC.
- H. R. Kokabi, J. Provost and G. Desgardin, Rev. Sci. Instrum., 1993, 64, 1549 CrossRef CAS.
- A. C. Larson and R. B. Von Dreele, General Structure Analysis System (GSAS), Los Alamos National Laboratory Report LA-UR-86-748, 2000, B. H. Toby, “EXPGUI, a graphical user interface for GSAS”, J. Appl. Crystallogr., 2001, 34, 210 CrossRef.
- A. M. Balagurov, J. Neutron Res., 2005, 16, 8 CrossRef.
- V. B. Zlokazov and V. V. Chernyshev, J. Appl. Crystallogr., 1992, 25, 447 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- G. N. Mazo, S. M. Kazakov, L. M. Kolchina, A. V. Morozov, S. Y. Istomin, N. V. Lyskov, A. A. Gippius and E. V. Antipov, J. Alloys Compd., 2015, 639, 381 CrossRef CAS.
- C. Tealdi, C. Ferrara, L. Malavasi, P. Mustarelli, C. Ritter, G. Chiodelli and Y. Diaz-Fernandez, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 8, 174118 CrossRef.
- F. Mauvy, C. Lalanne, J.-M. Bassat, J.-C. Grenier, H. Zhao, L. Huo and P. Stevens, J. Electrochem. Soc., 2006, 153, A1547 CrossRef CAS.
- A. Montenegro-Hernandez, J. Vega-Castillo, L. Mogni and A. Caneiro, Int. J. Hydrogen Energy, 2011, 36, 15704 CrossRef CAS.
- M. L. Medarde, J. Phys.: Condens. Matter, 1997, 9, 1679 CrossRef CAS.
- J. Gopalakrishnan, G. Colsmann and B. Reuter, J. Solid State Chem., 1977, 22, 145 CrossRef CAS.
- F. Tietz, Ionics, 1999, 5, 129 CrossRef CAS.
- J. W. Stevenson, T. R. Armstrong, L. R. Pederson, J. Li, C. A. Levinsohn and S. Baskaran, Solid State Ionics, 1998, 113–115, 571 CrossRef CAS.
- V. Vashook, J. Zosel, T.-L. Wen and U. Guth, Solid State Ionics, 2006, 177, 1827 CrossRef CAS.
- J. Yang, J. Cheng, Q. Jiang, Y. Wang, R. Wang and J. Gao, Int. J. Hydrogen Energy, 2012, 37, 1746 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Results of the refinement of the crystal structures using NPD data. See DOI: 10.1039/c6ra03099h |
| This journal is © The Royal Society of Chemistry 2016 |