
Catalytic transformation of glycerol to 1-propanol by combining zirconium phosphate and supported Ru catalysts†
Abstract
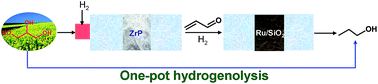
The one-pot hydrogenolysis of biomass-derived glycerol to 1-propanol has been investigated over sequential two-layer catalysts in a continuous-flow fixed-bed reactor. The zirconium phosphate layer was packed in the upper layer for dehydration of glycerol into acrolein and the supported Ru catalysts were in the second layer for the sequential hydrogenation of acrolein to 1-propanol. It was observed that the second layer catalyst with the strong acid sites would cause the formation of glycerol degradation products such as methanol, ethanol, methane and carbon dioxide etc., while 2%Ru/SiO2 with weak acid sites afforded the highest selectivity for 1-propanol. The sequential packing of zirconium phosphate and the 2%Ru/SiO2 catalytic system can give full glycerol conversions at 77% selectivity of 1-propanol, as well as exhibiting long-term stability (80 h). Carbonaceous deposits were a main reason for deactivation and the deactivated catalysts can be regenerated conveniently by calcinations in air. The present approach afforded an effective one-pot hydrogenolysis of glycerol to biopropanol, which could bring about the benign development of the biodiesel industry.
 Please wait while we load your content...
Please wait while we load your content...

