
Green synthesis of folic acid-conjugated gold nanoparticles with pectin as reducing/stabilizing agent for cancer theranostics†
Abstract
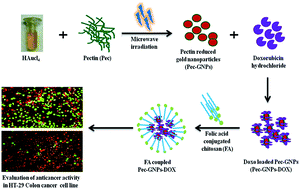
In the present study pectin, a natural polysaccharide was employed for the one pot aqueous synthesis of gold nanoparticles (GNPs). Pectin acted at the same time as both a reducing and stabilizing agent. The formation of pectin reduced GNPs (Pec-GNPs) was confirmed by using a UV-visible spectrophotometer, with a characteristic surface plasmon resonance (SPR) band at 527 nm. EDS analysis proved the presence of gold in the sample. The spherical morphology and crystalline nature of the Pec-GNPs was demonstrated by TEM analysis. The FTIR spectrum revealed the capping of pectin on the surface of the synthesised GNPs. Furthermore, the Pec-GNPs are found to be stable at different pH and electrolytic conditions. In vivo safety of the Pec-GNPs was established through zebra fish toxicity studies. The cationic drug doxorubicin was successfully loaded onto the synthesized anionic Pec-GNPs by an ionic complexation interaction. In vitro release studies confirmed the pH dependent sustained release of the doxorubicin. Doxorubicin loaded Pec-GNPs exhibited enhanced in vitro cytotoxicity on breast cancer cells compared to free doxorubicin, demonstrating that Pec-GNPs are efficient vehicles for the delivery of doxorubicin. Furthermore, chitosan coupled with folic acid (FA) was decorated with Pec-GNPs-DOX as a nanocarrier to improve the targeting and enhance the drug delivery to target cancer tissues by folic acid receptor-mediated endocytosis. It was concluded that the FA@Pec-GNPs-DOX were biocompatible and suitable for anti-cancer drug delivery, and were potentially promising as a new therapeutic system for cancer treatment.
 Please wait while we load your content...
Please wait while we load your content...

