
Carbonaceous layer interfaced TiO2/RGO hybrids with enhanced visible-light photocatalytic performance†
Jianfeng Xu,
Jiawei Tian,
Yuewei Zhang,
Ammara Riaz,
Yi Liu,
Mingjia Zhi,
Zhanglian Hong and
Chunmei Zhou*
State Key Laboratory of Silicon Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: cmzhou@zju.edu.cn
First published on 7th April 2016
Abstract
A facile solvothermal method combined with heat treatment was introduced to obtain enhanced photocatalytic activity of TiO2@C/RGO (reduced graphene oxide) photocatalyst under visible light. By adjusting the annealing temperature, the physical and chemical status of the interfacial carbonaceous layer could be successfully tuned. The carbonaceous layer with optimal thickness and degree of order obtained at 360 °C forms a good bridge between the TiO2 particles and RGO sheet. The resulting TiO2@C-360/RGO photocatalyst exhibited the best performance in the photodegradation of MO under visible light, benefiting from the ideal connection provided by the optimal carbonaceous layer and thus enhanced separation of activated electron–hole pairs at the interface. In addition, the photocatalytic activity was adjusted synchronously by controlling the thickness and degree of order of the carbonaceous layer in the TiO2@C/RGO hybrids. This work indicates the importance of interface engineering to obtain TiO2-based nanohybrids with high photocatalytic performance.
1 Introduction
In recent decades, TiO2-based nanohybrids have received increasing interest from scientists for their great potential in various industrial applications, such as photocatalysts, solar cells, and sensors.1–5 Owing to the concern to alleviate the deterioration of the environment by pollutants and the energy crisis and to ensure sustainable global development, as one of the most promising photocatalyst candidates, TiO2 hybrids are playing a more and more important role, with their superior properties, including chemical stability, non-toxicity, and low cost. However, the intrinsic band gap structure, ∼3.0–3.2 eV, only corresponding to the UV light spectrum region, strictly limits their practical use.6 Besides, the high recombination rate of photon activated electron–hole pairs is another main drawback seriously hindering its applications.7To solve these two big problems and thus improve the photocatalytic performance of TiO2 hybrids, researchers have proposed a lot of methods and successfully obtained TiO2 based photocatalysts with well-improved photocatalytic activity.8,9 Non-metal or metal element doping is one of the efficient ways found. Asahi et al. prepared N-doped TiO2 film which had good visible-light activity.10 Kim et al. obtained a solar light responsive TiO2 photocatalyst through co-doping with Fe and N.11 However, a higher electron–hole pair recombination rate usually follows doping. Noble metal modification is another powerful tool to extend the light absorption spectrum and reduce the recombination rate of the electron–hole pairs.12–15 Nevertheless, the high cost and relatively low stability of noble metals become new problems to be solved. Among a large amount of previous studies, photocatalysts modified with carbon materials exhibit superiority.16 Li et al., using glycerol as the carbon source, obtained a TiO2/C photocatalyst which had promoted photocatalytic activity.17 CdS coated with an amorphous carbon layer, synthesized by Hu et al., revealed the significance of the carbon in reducing the electron–hole recombination rate.18 Recently, TiO2/graphene hybrids have received great attention as graphene emerged as a new ideal 2-D carbon material with excellent properties, especially high carrier mobility and large surface area.19–21 Researchers have proved that the introduction of graphene in TiO2/graphene hybrids could greatly enhance the photocatalytic activity in visible light, which could be attributed to the large surface area, improved excited electron–hole separation and narrowed band gap of TiO2 hybrids.22–27
In spite of the incredible features of these hybrids, the photocatalytic activity is not always much higher than that of bare TiO2, because it is not easy to obtain ideal interfacial contact for TiO2/graphene hybrids, which may result in lower contact areas and weaker bonding forces between TiO2 and graphene. Thus these poor and noticeable interfaces would become recombination sites. Consequently, only few photo-induced electrons could be transferred through the interface, leading to a relatively low photocatalytic activity.
There have been few studies focusing on modification of the interface between TiO2 and graphene. In this work, a carbonaceous layer was employed to interface TiO2 particles and RGO (reduced graphene oxide) sheet, as an ideal charge carrier transmitter and effective stretcher from TiO2 to RGO. The thickness and degree of order of the carbonaceous layer was easily adjusted by post-annealing, which results in tunable photocatalytic activity for TiO2@C/RGO hybrids. This work exemplifies the importance of interface engineering in hybrid photocatalysts to optimize the photocatalytic activity and provides a new design strategy to fabricate ideal interfaces.
2 Experimental
2.1 Synthesis of TiO2@C samples
Titanium tetra-n-butoxide (TNB, 20 mL) (Sinopharm, CP) and 100 mL acetone were mixed in a quartz beaker which was placed in a 200 mL Teflon liner in an autoclave. De-ionized (DI) water (5 mL) was then added to the gap between the quartz beaker and the inner wall of the Teflon liner. TNB was hydrolyzed by the H2O evaporated from the gap when the autoclave was heated at 240 °C for 6 h. The precipitated powder was collected and washed with acetone and DI water several times and dried at 70 °C (named TiO2@C-pristine). It was then annealed at 180, 360 and 450 °C for 2 h and named TiO2@C-180, TiO2@C-360 and TiO2@C-450, respectively.2.2 Synthesis of TiO2@C/RGO hybrids
Home-made graphene oxide (GO, 10 mg) prepared by a modified Hummers' method and 200 mg as-prepared TiO2@C were dispersed in a solution containing 80 mL DI water and 40 mL ethanol under sonication and stirring, each for 2 h. The homogeneous suspension was kept at 120 °C for 3 h in a 200 mL autoclave. The precipitate was filtered and washed with ethanol and DI water, respectively, followed by drying at 70 °C overnight. The collected hybrids were named TiO2@C-pristine/RGO, TiO2@C-180/RGO, TiO2@C-360/RGO, and TiO2@C-450/RGO.2.3 Photocatalysis test
Photocatalyst (20 mg) was dispersed in 100 mL methyl orange (MO) solution (20 mg L−1). After stirring for 30 min in the dark, the suspension was illuminated under a halogen lamp (HQIBT, 400 W/D, OSRAM) using a filter with cut-off wavelength 420 nm to start photocatalytic degradation. After each hour, 5 mL suspension was collected and centrifuged at 8000 rpm to remove the photocatalyst. The supernatant was taken out for UV-vis absorption spectrum measurement. The relative concentration of residual MO solution (C/C0) at each hour was determined by the ratio of the intensity of the characteristic absorption peak at about 464 nm of the sample MO to that of the initial MO solution.2.4 Analysis of hydroxyl radical
In a typical process, 20 mL NaOH solution (2 mM) and 20 mL terephthalic acid (0.5 mM) were mixed in a quartz beaker and stirred for 30 min to form a homogeneous suspension. Then 20 mg TiO2@C/RGO sample was dispersed in the mixture with continuous stirring. The suspension was irradiated under visible light and readings were taken every 30 min. After removal of the residual photocatalyst by centrifugation at 8000 rpm for 10 min, the suspension was tested using the photoluminescence (PL) spectrum excited at 312 nm.2.5 Characterization
All samples were measured by powder X-ray diffraction (XRD), Rigaku, CuK = 0.15406 nm. Thermogravimetric analysis (TGA) curves were obtained by using an SDT-Q600 at a heating rate of 10 °C min−1. The morphology was analyzed by transmission electron microscopy (TEM, JEOL-1230, 120 kV) and high resolution transmission electron microscopy (HRTEM, Tecnai G2-F20-S-TWIN, FEI, 200 kV). Diffuse reflectance absorption spectra (DRS) were measured using a HITACHI U-4100 UV-vis spectrometer with an integrating sphere accessory and converted using the Kubelka–Munk function. Raman spectra were collected on a Renishaw Invia Raman spectrometer (532 nm, 50 mW). The photoluminescence (PL) emission spectra were recorded with a HITACHI F-4500 fluorescence spectrophotometer. PL decay curves were measured by using an Edinburgh FLS920 fluorescence spectrometer with 375 nm and 980 nm pulsed lasers.3 Results and discussion
3.1 Characterization of TiO2@C samples
The thermal stability of the as-prepared TiO2@C-pristine sample (made from organic Ti precursor) was analyzed by TGA, and compared with commercial TiO2 (made from inorganic Ti precursor), as shown in Fig. 1. The TGA curve of TiO2@C-pristine shows three weight-loss regions. A dramatic loss of weight can be observed before 180 °C, and the weight loss is ca. 17%, which can be ascribed to the dehydration and evaporation of volatile carbonaceous organic species. The second stage is found from 180 °C to 360 °C with a ca. 7% weight loss. In this region, the weight decreases at a stable speed which may be attributed to the combustion and carbonization of the remaining organic species. The third stage is between 360 °C and 450°, with a slight mass decrease, only ca. 2%, which is mainly caused by the further oxidation of the amorphous carbon layer formed by carbonization under air atmosphere. By comparison with the TGA curve of commercial anatase TiO2 particles, a more than 12% of weight loss for TiO2@C sample can be observed, which further confirms the existence of organic species around the TiO2 particles. The evolution of the incorporated carbonaceous layer for the TiO2 precursor is almost complete at 450 °C because there is no obvious mass decrease above this temperature. Based on the TGA analysis, post-annealing was selected as the main tool to modify the surface state of TiO2@C samples, and TiO2@C samples with various surface states were obtained by heat treatment at different temperatures (180, 360, and 450 °C). Raman spectroscopy, XRD and TEM were used to explore the quality and degree of order of the carbonaceous layer, phase structure and crystallite sizes of the TiO2@C samples. | ||
| Fig. 1 TGA curve of TiO2 samples derived from hydrothermal synthesis and commercial TiO2. | ||
Fig. 2 depicts the micro Raman spectra of TiO2@C samples with D band at 1350 cm−1 and G band at 1580 cm−1, which are related to the vibration of carbon atoms in disordered carbon structure or defect sites and in-plane vibration of sp2-bonded carbon atoms, respectively.28 The ratio of intensities of two peaks (ID/IG) is normally used as a measurement to represent the amount of defects and the degree of disorder inside carbon materials.29,30 In this way, a less defective degree of order would be translated into a lower ratio of ID/IG in the Raman spectra. As shown in Fig. 2 and Table 1, the values of ID/IG indicate that TiO2@C-180 and TiO2@C-360 have a relatively ordered carbonaceous layer, 0.99 and 0.99, whereas those of TiO2@C-pristine and TiO2@C-450, with higher degree of disorder, are 3.98 and 1.12, respectively. This is also consistent with the results of TGA analysis as more defects in the organic species exist on the surface of TiO2@C-pristine and no complete carbon layer is left on the surface of TiO2@C-450 sample owing to too much oxidation.
 | ||
| Fig. 2 Raman spectra of TiO2@C samples using a 532 nm laser with a power of 50 MW. | ||
| Samples | Particle sizea | ID/IGb | Band gapc (eV) | Degradationd |
|---|---|---|---|---|
| a The average particle sizes of TiO2@C samples were calculated by the Scherrer formula based on the XRD patterns (Fig. S1) and those of TiO2@C/RGO were calculated based on TEM images (Fig. 3).b Intensity ratio of peak D and peak G in Raman spectrum (Fig. 2).c The band gap was obtained based on the UV-vis absorption spectra (Fig. 5).d The photodecomposition was carried out with 20 mg photocatalyst and 20 mg L−1 MO under visible light (>420 nm) for 4 h. | ||||
| TiO2@C-pristine | 15.1 | 3.98 | 3.20 | ∼0 |
| TiO2@C-180 | 15.2 | 0.99 | 3.20 | ∼0 |
| TiO2@C-360 | 15.2 | 0.99 | 3.25 | ∼0 |
| TiO2@C-450 | 15.6 | 1.12 | 3.20 | ∼0 |
| TiO2@C-pristine/RGO | 16.3 | — | 2.8 | 23% |
| TiO2@C-180/RGO | 16.1 | — | 2.8 | 59% |
| TiO2@C-360/RGO | 16.0 | — | 2.8 | 66% |
| TiO2@C-450/RGO | 15.8 | — | 2.8 | 18% |
Powder XRD was used to investigate the phase structure and crystallite sizes of the TiO2@C samples in Fig. S1.† The XRD patterns are analogous to the anatase phase (JCPDs 21-1272) in which sharp peaks indicate the high crystallinity of TiO2. No remarkable variation of the full width at half-maximum of the TiO2 (101) plane indicates no significant crystalline growth of TiO2 during the post-annealing process, which is consistent with the average sizes shown in Table 1 calculated by the Scherrer formula. This result means the particle size and crystallinity will have almost the same effect on the photoactivity of the four typical TiO2@C/RGO samples. The commercial TiO2 powder used in TGA analysis was also anatase, as shown in Fig. S1.† In addition, the HRTEM image of TiO2@C-360 in Fig. 3(a) directly displays the amorphous thin carbonaceous layer uniformly formed on the surface of TiO2. And the lattice fringe of TiO2 is 0.35 nm for the nanoparticles, matching well the typical distance for the anatase (101) plane.
 | ||
| Fig. 3 HRTEM images of TiO2@C-360 (a) and TiO2@C-360/RGO (b) and TEM images of TiO2@C/RGO hybrids (c–f). | ||
TiO2@C samples with an amorphous carbonaceous layer were successfully prepared by using the organic Ti precursor. As a result of post-annealing at different temperatures, TiO2@C samples with various surface chemical states and degrees of order were obtained. And the annealing process has little effect on the grain size and crystallinity of TiO2.
3.2 Characterization of TiO2@C/RGO photocatalysts
To extend the absorption spectrum and explore the effect of the interfacial carbonaceous layer on activated electron–hole separation, the as-prepared TiO2@C samples were combined with reduced graphene oxides (RGO) by a solvothermal process; the products were named TiO2@C-pristine/RGO, TiO2@C-180/RGO, TiO2@C-360/RGO and TiO2@C-450/RGO. The whole synthesis flow diagram is illustrated in Scheme 1. After the second solvothermal process, the GO was reduced to RGO, which could be proved by XRD patterns of GO and RGO as shown in Fig. S2.† The (001) peak of GO disappeared and a strong (002) peak of RGO formed at about 25°. | ||
| Scheme 1 Synthesis of TiO2@C/RGO photocatalyst. | ||
The low-resolution transmission electron microscopy (TEM) images in Fig. 3(c–f) show the morphology of the TiO2@C/RGO hybrids. TiO2 nanocrystals of less than 20 nm are well dispersed on the two-dimension RGO sheet and the average sizes of particles seen in the TEM images of the four samples were calculated by Nano Measurer software, as shown in Table 1 and Fig. S3.† It is apparent that the average sizes from TEM slightly decrease with temperature. The reason could be attributed to the amorphous carbonaceous layer being gradually removed by increasing temperature. The HRTEM image of TiO2@C-360/RGO in Fig. 3(b) directly shows that the amorphous carbonaceous layer still exists on the surface of TiO2 and the particles are uniformly adhered to RGO. The lattice fringe of TiO2 is also the same as for the TiO2@C-360. In this way, TiO2@C/RGO hybrids with a tunable carbonaceous layer were successfully obtained.
Fig. 4 shows the UV-visible absorption properties of TiO2@C and TiO2@C/RGO samples. An obvious red-shift in absorption edge and strong absorption in the visible range can be observed for TiO2@C/RGO hybrids in Fig. 4(a) because of the introduction of RGO. Photographs of TiO2@C/RGO hybrids are shown in Fig. S4,† in which all the samples are black-gray, suggesting good sensitivity to visible light. Fig. 4(b) shows plots of the Kubelka–Munk remission function (relationship of [αhν]1/2 to photon energy). The band gaps of TiO2@C/RGO hybrids are all significantly reduced to 2.80 eV which means the existence of carbonaceous layers with different thicknesses and degrees of order has no effect on the band gap for these hybrids.
 | ||
| Fig. 4 UV-vis absorption spectra of TiO2@C and TiO2@C/RGO samples. | ||
X-ray photoelectron spectroscopy (XPS) measurements were carried out to check the chemical status of TiO2@C/RGO hybrids. The fine spectrum of C1s of TiO2@C/RGO is shown in Fig. 5. The peaks at around 284.7, 286.0 and 288.3 eV correspond to C–C, C–O–R and Ti–O–C bonds.31 There is no peak found at 282 eV, suggesting the absence of carbon doping during the annealing process.32 The C–C bonds in TiO2@C/RGO hybrids are found in three regions: (1) the surface carbon of TiO2, (2) the RGO and (3) the bond between surface carbon and RGO. The proportions of C–C bonds in TiO2@C-pristine/RGO in Fig. 5(a) and TiO2@C-180/RGO in Fig. 5(b) are 84.2% and 81.2%, respectively. As discussed for TG analysis and Raman spectra, a thick carbonaceous layer with more organic species exists in the TiO2@C-pristine/RGO hybrid, while the thinner carbonaceous layer with lower degree of disorder in TiO2@C-180/RGO contributes to a lower proportion of C–C bonds, which may induce a lower recombination rate at the interface. The high proportion of C–C bonds in TiO2@C-360/RGO seen in Fig. 5(c), 90.7%, is assumed to result from strong conjunction between the surface carbon shell around TiO2 and the RGO, and is much higher than that of TiO2@C-450/RGO, 79.2%. This excellent bridging connection induced by the optimal carbonaceous layer in TiO2@C-360/RGO hybrids may provide more charge transfer paths during the photocatalysis.
 | ||
| Fig. 5 XPS of C1s in TiO2@C/RGO sample: TiO2@C-pristine/RGO, TiO2@C-180/RGO, TiO2@C-360/RGO and TiO2@C-450/RGO. | ||
In this way, TiO2@C/RGO photocatalysts with a tunable carbonaceous layer were obtained. The existence of the carbonaceous layer has little effect on the band gap of TiO2@C/RGO photocatalysts. However, the interfaced carbonaceous layer may contribute to the highly efficient separation of activated electron–hole pairs.
3.3 Photocatalytic performance of TiO2@C/RGO photocatalysts
To evaluate the photocatalytic activity and explore the synergetic effect of the carbonaceous layer, the decomposition of methylene orange (MO) solution (20 mg L−1) under visible light (>420 nm) with 20 mg photocatalyst was used as a probe. As shown in Fig. 6, the decomposition of MO followed the order: TiO2@C-450/RGO < TiO2@C-pristine/RGO < TiO2@C-180/RGO < TiO2@C-360/RGO, with the amount of MO decomposed over the course of 4 h: 18%, 23%, 59% and 66%, respectively. Obviously, the different states of carbonaceous interface layer modified by annealing greatly influenced the photocatalytic activity.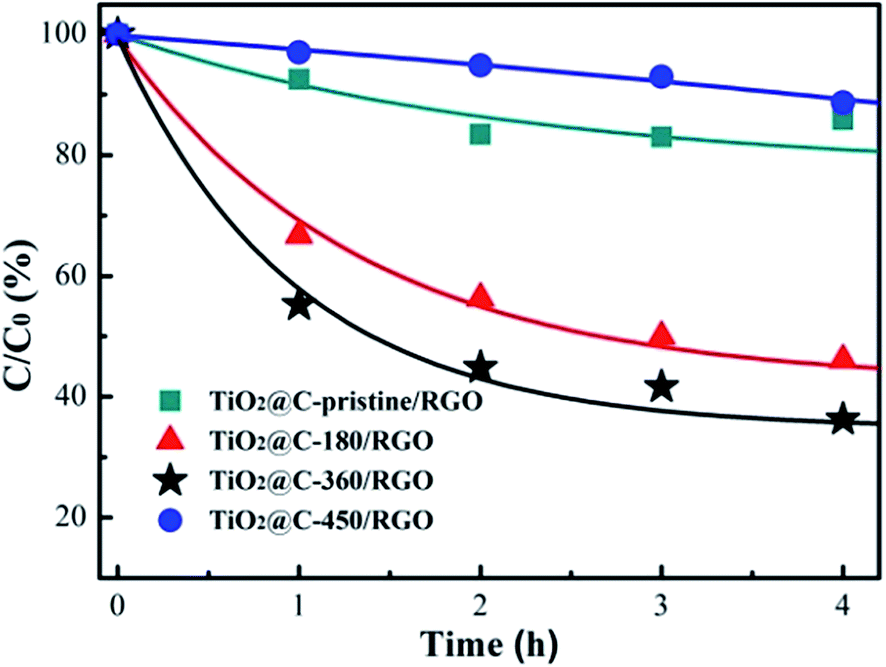 | ||
| Fig. 6 Photocatalytic degradation curves of MO in the presence of TiO2@C/RGO photocatalysts under visible light (>420 nm). | ||
PL spectra excited at 420 nm were used to analyse the actual effect of the amorphous carbonaceous layer on the optical properties of the photocatalysts in visible light. As shown in Fig. 7, TiO2@C-360/RGO exhibits the weakest emission peak, followed by TiO2@C-180/RGO, TiO2@C-pristine/RGO and TiO2@C-450/RGO. The emission peaks appearing between 430 nm and 460 nm arise from continuous excitonic e−–h+ recombination.33,34 It is worthy of note that the result is in good agreement with the degree of order of the samples. TiO2@C-360/RGO and TiO2@C-180/RGO, having carbonaceous layers with high degrees of order and fewer defects, allow efficient transfer of photo-induced electrons to RGO. In addition, the emission intensity is also closely related to the thickness of the carbonaceous layer. TiO2@C-360/RGO has a thinner carbonaceous layer with optimized degree of order compared with TiO2@C-180/RGO, so that the connection of TiO2 particles and RGO by C–C bonds in the TiO2@C-360/RGO photocatalyst is much better for the efficient separation of activated electron–hole pairs.35 TiO2@C-pristine/RGO possesses more surface defects and carbonaceous organic species, which will provide numerous recombination sites and inhibit good contact with graphene. Similarly, as there is no efficient transmission path for TiO2@C-450/RGO to deliver the carriers to RGO, attributed to the poor connection between TiO2 and RGO, the emission peak is much higher. These results indicate that an optimized thin and ordered carbonaceous layer is very important to bridge the nanoparticles and graphene to enhance the charge transfer at the interface of the hybrids and thus improve the photocatalytic performance.
 | ||
| Fig. 7 PL emission peaks after excitation at 420 nm for TiO2@C/RGO photocatalysts. | ||
More supporting evidence is provided by the decay time of the samples shown in Fig. 8, which gives a direct view of the process of electron diffusion under laser irradiation at 375 nm. The signal is collected at 440 nm and reveals the lifetime of the samples in this order: TiO2@C-pristine/RGO < TiO2@C-450/RGO < TiO2@C-180/RGO < TiO2@C-360/RGO.
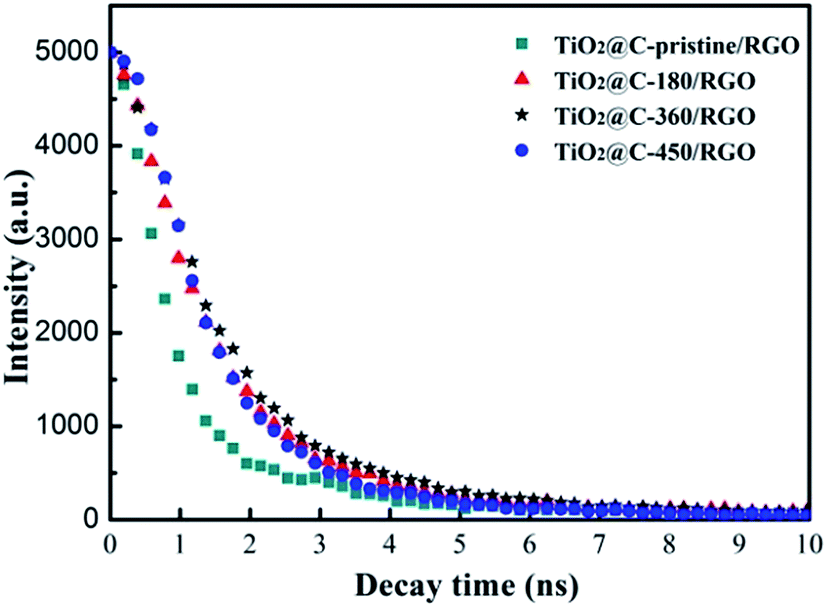 | ||
| Fig. 8 PL decay curves of TiO2@C/RGO photocatalysts. | ||
To evaluate the recyclability of the photocatalyst with the best performance, the sample was collected by centrifugation at 8000 rpm after the first run of the photodegradation test. The collected material was dispersed in water for desorption for 30 min and then centrifuged at 8000 rpm again. This operation was carried out 3 times. After weighing, the lost mass, a few milligrams, was made up and the photocatalysis test was repeated as previously. As shown in Fig. 9, the TiO2@C-360/RGO photocatalyst possesses stable recycling ability. After the test had been carried out for another two cycles, the degradation rate of TiO2@C-360/RGO photocatalyst was maintained at 64% in 4 h under visible light, suggesting this photocatalyst is capable of being used for long-term runs.
 | ||
| Fig. 9 Recycling test of TiO2@C-360/RGO for photocatalytic degradation of MO under visible light. | ||
Above all, the photocatalytic activity of TiO2@C/RGO could be optimized by tuning the interfacial carbonaceous layer. The TiO2@C-360/RGO hybrid, with a relatively thin and ordered carbonaceous layer, exhibits the best charge transfer power and stable recycling ability, which is in good agreement with the photocatalytic decomposition results. Besides, the TiO2@C-360/RGO photocatalyst also exhibits much higher photocatalytic activity than P25/RGO prepared under the same conditions, as shown in Fig. S5.†
3.4 Photocatalysis mechanism
When TiO2 particles are combined with RGO, the band gap of the obtained hybrid can be lowered to visible as reported in a previous study.23 However, the charge separation at the interface between TiO2 and RGO is still a considerable factor affecting the photocatalytic activity of the hybrid. When the photocatalyst is irradiated under visible light, TiO2 could produce activated electron–hole pairs. The modified carbonaceous layer of TiO2@C-360/RGO with optimal degree of order and optimal thickness could provide an efficient separation path for the electron–hole pairs. Thus the activated electron could be transferred to RGO in time to form oxygen radicals and the remaining hole could be used to form hydroxyl radicals. Fig. 10 shows the PL emission spectra of terephthalic acid with TiO2@C/RGO sample under visible light irradiation. Clearly, a PL peak at about 425 nm is observed when the suspension is illuminated by visible light, and the intensity increases with time, indicating that ˙OH is produced during the photocatalytic reaction. Moreover, PL intensity for TiO2@C-360/RGO in Fig. 10(a) is much stronger than that for TiO2@C-450/RGO in Fig. 10(b) at the same time, which means the amount of ˙OH radicals produced is much more on TiO2@C-360/RGO than TiO2@C-450/RGO. The better interfacial structure induced by the optimal carbonaceous layer improves the separation of activated electron–hole pairs and promotes the formation of ˙OH radicals and ˙O2− radicals, contributing to enhanced photocatalytic activity. Finally, the MO pollutant will react with oxygen radicals and hydroxyl radicals to form water and carbon dioxide, as illustrated in the mechanism shown in Scheme 2. | ||
| Fig. 10 PL emission spectra of terephthalic acid with TiO2@C/RGO sample under visible light irradiation excited at 312 nm: (a) TiO2@C-360/RGO; (b) TiO2@C-450/RGO. | ||
 | ||
| Scheme 2 Mechanism of the photocatalytic process on TiO2@C/RGO. | ||
4 Conclusions
A novel TiO2@C/RGO photocatalyst was synthesized in this study. The carbonaceous layer modified TiO2 nanoparticles were anchored on RGO sheets by a solvothermal process. The hybrid with moderate post-annealing, heat-treatment at 360 °C, exhibited the best visible-light photocatalytic activity, due to the sufficient contact and enhanced charge transfer caused by the thin and ordered carbonaceous layer at the interface between TiO2 and RGO. The modification of the interfaced carbonaceous layer in these hybrids has been proved to be powerful in improving the photocatalytic activity. This result of our study indicates the importance of interface engineering to achieve highly active photocatalysts and better understand the photoactivity mechanism of semiconductor/graphene hybrids.Acknowledgements
This work was supported partially by National Natural Science Foundation of China (21301153 and 21503187).References
- J. T. Li and N. Q. Wu, Catal. Sci. Technol., 2015, 5, 1360–1384 CAS.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41, 782–796 RSC.
- Y. B. Tang, C. S. Lee, J. Xu, Z. T. Liu, Z. H. Chen and Z. B. He, ACS Nano, 2010, 4, 3482–3488 CrossRef CAS PubMed.
- D. H. Wang, D. W. Choi, J. Li, Z. G. Yang, Z. M. Nie and R. Kou, ACS Nano, 2009, 3, 907–914 CrossRef CAS PubMed.
- M. M. Gao, L. L. Zhu, W. L. Ong, J. Wang and G. W. Ho, Catal. Sci. Technol., 2015, 5, 4703–4726 CAS.
- A. Fujishima, X. T. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- L. Run, J. E. Niall and V. P. Oleg, J. Am. Chem. Soc., 2012, 134, 14238–14248 CrossRef PubMed.
- A. Ryu, J. Photochem. Photobiol., C, 2010, 11, 179–209 CrossRef.
- C. Peng, X. Yang, Y. Li, H. Yu, H. Wang and F. Peng, ACS Appl. Mater. Interfaces, 2016, 8, 6051–6060 CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- T. H. Kim, V. R. González, G. Gyawalia, S. H. Cho, T. Sekinoc and S. W. Lee, Catal. Today, 2013, 212, 75–80 CrossRef CAS.
- E. Kowalska, R. Abea and B. Ohtani, Chem. Commun., 2009, 241–243 RSC.
- Z. W. Liu, W. B. Hou, P. Pavaskar, M. Aykol and S. B. Cronin, Nano Lett., 2011, 11, 1111–1116 CrossRef CAS PubMed.
- Y. Tian and T. Tatsuma, J. Am. Chem. Soc., 2005, 127, 7632–7637 CrossRef CAS PubMed.
- J. T. Li, S. K. Cushing, P. Zheng, F. K. Meng, D. Chu and N. Q. Wu, Nat. Commun., 2013, 4, 2095 Search PubMed.
- R. Leary and A. Westwood, Carbon, 2011, 49, 741–772 CrossRef CAS.
- M. Li, S. F. Zhou, Y. W. Zhang, G. Q. Chen and Z. L. Hong, Appl. Surf. Sci., 2008, 254, 3762–3766 CrossRef CAS.
- Y. Hu, X. H. Gao, L. Yu, Y. R. Wang, J. Q. Ning, S. J. Xu and X. W. Lou, Angew. Chem., Int. Ed., 2013, 52, 5636–5639 CrossRef CAS PubMed.
- X. Q. An and J. Yu, RSC Adv., 2011, 1, 1426–1434 RSC.
- N. Zhang, Y. H. Zhang and Y. J. Xu, Nanoscale, 2012, 4, 5792–5813 RSC.
- W. G. Tu, Y. Zhou and Z. G. Zou, Adv. Funct. Mater., 2013, 23, 4996–5008 CrossRef CAS.
- C. Chen, W. M. Cai, M. C. Long, B. X. Zhou, Y. H. Wu, D. Y. Wu and Y. J. Feng, ACS Nano, 2010, 4, 6425–6432 CrossRef CAS PubMed.
- B. Bukowski and N. A. Deskins, Phys. Chem. Chem. Phys., 2015, 17, 29734–29746 RSC.
- J. S. Lee, K. H. You and C. B. Park, Adv. Mater., 2012, 24, 1084–1088 CrossRef CAS PubMed.
- J. A. J. Du, Y. H. Ng, N. J. Bell, Z. H. Zhu, R. Amal and S. C. Smith, J. Phys. Chem. Lett., 2011, 2, 894–899 CrossRef PubMed.
- M. C. Long, Y. L. Qin, C. Chen, X. Y. Guo, B. H. Tan and W. M. Cai, J. Phys. Chem. C, 2013, 117, 16734–16741 CAS.
- H. Kim, S. Kim, J. K. Kang and W. Choi, J. Catal., 2014, 309, 49–57 CrossRef CAS.
- N. R. Khalid, Z. L. Hong, E. Ahmed, Y. W. Zhang, H. Chan and M. Ahmad, Appl. Surf. Sci., 2012, 258, 5827–5834 CrossRef CAS.
- A. T. T. Koh, Y. M. Foong and D. H. C. Chua, Appl. Phys. Lett., 2010, 97, 114102 CrossRef.
- Z. H. Ni, H. M. Fan, X. F. Fan, H. M. Wang, Z. Zheng, Y. P. Feng, Y. H. Wu and Z. X. Shen, J. Raman Spectrosc., 2007, 38, 1449–1453 CrossRef CAS.
- L. Chen, F. Chen, Y. Shi and J. Zhang, J. Phys. Chem. C, 2012, 116, 8579–8586 CAS.
- F. Dong, H. Wang and Z. Wu, J. Phys. Chem. C, 2009, 113, 16717–16723 CAS.
- A. A. Lsmail, R. A. Geioushy, H. Bouzid, S. A. Al-Sayari, A. Al-Hajry and D. W. Bahnemann, Appl. Catal., B, 2013, 129, 62–70 CrossRef.
- L. Kernazhitsky, V. Shymanovska, T. Gavrilko, V. Naumov, L. Fedorenko, V. Kshnyakin and J. Baran, J. Lumin., 2014, 16, 199–204 CrossRef.
- A. J. Cheah, W. S. Chiu, P. S. Khiew, H. Nakajima, T. Saisopa, P. Songsiriritthigul, S. Radimane and M. A. A. Hamide, Catal. Sci. Technol., 2015, 5, 4133–4143 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02431a |
| This journal is © The Royal Society of Chemistry 2016 |