
A mesoporous aluminosilicate prepared by simply coating fibrous γ-AlOOH on the external surface of SBA-15 for catalytic hydrocarbon cracking†
Zongbo Shi,
Yejun Guan*,
Peng Wu and
Mingyuan He*
Shanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, North Zhongshan Rd. 3663, Shanghai 200062, China. E-mail: yjguan@chem.ecnu.edu.cn; hemingyuan@126.com
First published on 18th April 2016
Abstract
A binary SiO2/Al2O3 composite with fibrous γ-alumina coating on the external surface of SBA-15 has been synthesized by simply mixing SBA-15 with fibrous boehmite sol, followed by aging and calcination. The textural and acidic properties of the resultant material were characterized by various techniques, i.e. powder X-ray diffraction (XRD), scanning electron microscopy (SEM), infrared (IR) spectroscopy, N2 adsorption/desorption, nuclear magnetic resonance (NMR) spectroscopy, and temperature programmed desorption of ammonia and ethanol (NH3-TPD and EtOH-TPD). Coating fibrous γ-alumina stabilized the SBA-15 structure against collapse under critical steaming conditions (800 °C and 4 h) allowing for the facile synthesis of mesoporous aluminosilicate. The migration of aluminum and silicon between the alumina and SBA-15 zones led to the formation of Al–OH–Si with weak acidity which served as the active sites with excellent catalytic performance in the cracking of 1,3,5-triisopropyl benzene (TIPB).
Introduction
Fluidized catalytic cracking (FCC) aims to convert heavy fractions in vacuum distillate oil to clean liquid fuels with small molecular sizes.1 The major active components of FCC catalysts are FAU- or MFI-type zeolites that exhibit strong acidity and excellent hydrothermal stability. Due to an increasing demand to process heavier residues (boiling point > 400 °C, carbon number > 20), it is highly desirable that these bulky molecules could undergo pre-cracking on acid sites which locate on the external surface provided by the matrix.2 In this regard, developing FCC catalysts with high accessibility and active matrices such as nonzeolitic aluminosilicates, also called amorphous silicon alumina (ASA) with moderate acidity and mesoporosity are quite promising.3,4 Moreover, the matrices should be resistant to the severe deactivation conditions of the practical regeneration, with temperatures above 700 °C or even higher in steam.5–7Recent years have witnessed substantial progress in preparing active matrices such as Al2O3, SiO2 or binary SiO2/Al2O3 composites with improved textural properties, enhanced surface acidity, greater hydrothermal stability.8–10 Their practical applications used as a matrix have been widely explored in many reactions such as hydrocarbon cracking, propylene oligomerization, lube dewaxing and the conversion of methanol to hydrocarbons.6,11,12 Ishihara and coworkers reported that hierarchical ZSM-5 containing mesoporous silica–aluminas showed not only higher conversion of VGO but also higher yields of gasoline compared with that of single ZSM-5.13 Zhuo et al. prepared sprayed FCC catalyst by using a mixture of silica sol and alumina sol as the binder and active component, which exhibited improved heavy oil conversion and transport fuels selectivity.14 The enhanced catalytic activity is likely related to the mesopores and the transfer of aluminium and/or silicon species from the matrix to the zeolite phase.15
Natural clays can be directly used as SiO2/Al2O3 source and have been widely studied.16 Recently, it is found that coating γ-Al2O3 on the surface of SiO2 brings several benefits: the crystalline alumina-containing aluminosilicate usually possesses high hydrothermal stability in surface area, partly because the γ-Al2O3 was thermal stable in a range of temperature.17,18 Jin et al. demonstrated that SiO2@Al2O3 powders possess higher thermal conductivity than SiO2 powder, which helped to delay catalyst from overheating. On the other hand the SiO2@Al2O3 powders were of low surface area and showed limited catalytic activity.19,20 It is highly desirable to synthesize the active SiO2–Al2O3 matrix with high surface area. Yu and coworkers synthesized SiO2@Al2O3 with the surface area of 653 m2 g−1 using electrostatic attraction and a heterogeneous nucleation strategy, where the microspheres SiO2 with surface area of 999 m2 g−1 were used as the core.21
Here we reported a facile synthesis of a binary SiO2/Al2O3 composite by coating fibrous γ-alumina on the external surface of SBA-15. This simple approach combined electrostatic attraction strategy, aging and calcination, resulting in the formation of weak acid sties by the migration of silicon and aluminum species inside the matrix. Thus obtained binary SiO2/Al2O3 composite can efficiently catalyze the cracking of TIPB to diisopropyl benzene, cumene, benzene and propylene.
Experimental
Chemicals
Non-ionic triblock co-polymer Pluronic P123 (EO20PO70EO20, molecular weight = 5800) were purchased from J&K Chemical Ltd. HCl (37 wt%), AlCl3·6H2O, Al(NO3)3·9H2O, NaOH, tetraethylorthosilicate (TEOS), aluminum isopropoxide, hexane all in AR grade, were supplied by Sinopharm Chemical Reagent Co. Ltd. The chemicals were used directly without further purification.Preparation of binary SiO2/Al2O3 composite
Binary SiO2/Al2O3 composite was synthesized by mixing SBA-15 with fibrous boehmite sol, followed by aging and calcination. First SBA-15 was synthesized according to the procedure reported by Zhao et al.22 Typically, 2.0 g Pluronic P123 was dissolved in 60 mL of 2.0 M HCl and 15 mL of distilled water, to which 4.3 g of TEOS was added. The resulting mixture was continuously stirred at 40 °C for 24 h, and then transferred into a Teflon-lined stainless steel autoclave and aged at 100 °C for 24 h. After crystallization, the solid product was filtered, washed with deionized water repeatedly, and dried overnight at 100 °C in air. The as-synthesized sample was calcined at 550 °C for 6 h with a heating rate of 2 °C min−1.Then fibrous boehmite sol (AlOOH sol), fibrous boehmite (AlOOH) and fibrous alumina (Al2O3) were synthesized according to the procedure reported by Deng et al.23 Typically, 20 mmol of AlCl3·6H2O was dissolved into 40 mL water under stirring at room temperature, to which 60 mL of 1 M NaOH was added dropwise. Then 1 M HCl was added to adjust the pH to ∼4. The obtained suspension was transferred into a Teflon-lined stainless steel autoclave and aged at 175 °C for 12 h. The resulting suspension was named as AlOOH sol. The AlOOH was collected by centrifugation of AlOOH sol, and dried at 100 °C overnight. The Al2O3 was obtained by calcination of AlOOH at 550 °C for 6 h with a heating rate of 2 °C min−1.
SBA-15/fibrous boehmite composite (SBA–AlOOH) and SBA-15/fibrous alumina composite (SBA–Al2O3) were obtained as following: 20 mmol SBA-15 was added into the fibrous boehmite sol (containing 20 mmol Al2O3) under continuously stirred at room temperature for 2 h. The resulting suspension was transferred into a Teflon-lined stainless steel autoclave and aged at 150 °C for 4 h, then cooled down naturally. The SBA–AlOOH product was collected by filtration, washed with distilled water and dried at 80 °C overnight. The SBA–Al2O3 was obtained by calcination of SBA–AlOOH at 550 °C for 6 h with a heating rate of 2 °C min−1.
For comparison, alumina incorporated SBA-15 (SBA/Al2O3-imp) with the same SiO2/Al2O3 ratio was also prepared by impregnation according to the reported procedure.24 The mixture of 10 mmol Al(NO3)3·9H2O and 10 mmol SBA-15 was ground for 0.5 h in an agate mortar. Then the mixture was calcined at 550 °C for 6 h with a heating rate of 2 °C min−1.
To compare the hydrothermal stability of the aluminosilicates, the steamed samples, namely steamed Al2O3, steamed SBA–Al2O3 and steamed SBA/Al2O3-imp were obtained by calcining Al2O3, SBA–Al2O3 and SBA/Al2O3-imp at 800 °C in 100% steam for 4 h, respectively.
Characterization methods
Powder X-ray diffraction (XRD) patterns were collected on a Rigaku-Ultima diffractometer with a nickel filter using a Cu Kα radiation source (λ = 0.15418 nm, 35 kV, 25 mA) in the 2θ range from 0.5–5° (0.5° min−1) and 10–80° (10° min−1). Scanning electron microscopy (SEM) was performed directly without spraying gold on a Hitachi S-4800 microscope. Infrared (IR) spectra were recorded on a Nicolet Fourier transform infrared spectrometer (NEXUS 670). The self-supported wafers without mixing with KBr (about 10 mg, ∅ 1 cm) were pretreated in the IR cell under vacuum at 350 °C for 1 h and then the IR spectra of OH group were recorded. After the samples were cooled down to room temperature, pyridine vapor was dosed into the IR cell. The physically adsorbed pyridine was removed by evacuation at 150 °C. N2 adsorption–desorption isotherms were measured at −196 °C on a Quanta chrome Autosorb-3B volumetric adsorption analyzer. Before the measurements, the samples were degassed at 300 °C for 6 h. The BET (Brunauer–Emmett–Teller) specific surface area was calculated using adsorption data acquired at a relative pressure (P/P0) range of 0.05–0.30 and the total pore volume was determined from the amount adsorbed at P/P0 of about 0.99. The pore size distribution (PSD) curves were calculated from the analysis of adsorption isotherm branches using the Barrett–Joyner–Halenda (BJH) algorithm. The bulk Si, Al and Na contents were determined by inductively coupled plasma emission spectrometry (ICP) on a Thermo IRIS Intrepid II XSP atomic emission spectrometer. The nuclear magnetic resonance spectroscopy of 27Al (27Al NMR) were measured on a VARIAN VNMRS 400WB NMR spectrometer.Catalytic cracking of triisopropyl benzene
A stainless steel tube micro reactor with an inner diameter of 10 mm was used for the cracking experiments. The samples were all ammonium exchanged before catalytic test. 1.0 g of the catalyst particles (40–60 mesh size) were loaded and in situ activated at 400 °C for 2 h under a 40 sccm flow of N2. Afterwards, N2 was switched off and 1.506 g TIPB was injected into the carburetor with the flow rate of 1.29 g min−1. TIPB was immediately vaporized and flowed into the reactor. The reaction products were cooled with an ice-water bath. After the reaction, the N2 was switched on for purging the residual products, with a flow rate of 40 sccm for 10 min. The weight of liquid products and the volume of gas products were recorded. The liquid products were analyzed by an offline gas chromatograph (GC) equipped with a flame ionization detector (FID) and a HP-Plot Q column (∅ 0.32 mm and 50 m length). The gas products were analyzed by an online GC equipped with a thermal conductivity detector (TCD), a FID and a DM-alumina column (∅ 0.53 mm and 50 m length). Almost no TIPB thermal cracking was observed at 400 °C without catalyst.The refractory property of the coke formed on each catalyst was studied via TPO technique.25 In a typical experiment, 50 mg sample was pretreated at 400 °C under 25 sccm flow of helium for 1 h and cooled to 100 °C. Afterwards helium was switched off and 40 sccm flow of 20 vol% O2/N2 was passed through the coked catalyst at 100 °C for 0.5 h. Then the sample was heated from 100 °C to 800 °C with ramp rate of 10 °C min−1. The exit stream was passed through a CuO/Cu2O catalyst bed held at 500 °C which converted the CO formed and any desorbing hydrocarbons to CO2 and H2O.26 After removing the water by passing through a neutral desiccant, the CO2 was measured by a TCD monitor and the amount of the coke was calibrated with commercial carbon as reference.
Results and discussion
Characterization of SBA–Al2O3 materials
Fig. 1 displays the X-ray diffraction patterns of the SBA-15, Al2O3 and SBA–Al2O3 samples. The SBA-15 sample illustrated three well resolved peaks at 2θ = 1.02°, 1.74°, 2.00° (Fig. 1a, left), corresponding to the (100), (110) and (200) diffractions of a 2D hexagonal space group (P6mm), respectively. The unit cells calculated by the XRD pattern are ∼10 nm. For SBA–Al2O3, similar diffraction peaks due to SBA-15 were observed but with lower intensity (Fig. 1c). New peaks assigned to γ-alumina (JCPDS card number 04-0880) were found. | ||
| Fig. 1 Small-(left) and wide-(right) angle XRD patterns of the as synthesized SBA-15 (a), Al2O3 (b), and SBA–Al2O3 (c). | ||
Fig. 2 shows the typical SEM images of the resulting materials. SBA-15 particles (Fig. 2a) exhibit hexagonal cylinder like morphologies, which is consistent with the XRD results. The fibrous boehmite (AlOOH) particles (Fig. 2b) had uniform fibrous-like morphology, with length in 0.5–1 μm and diameter in 6–9 nm. Fig. 2c shows the morphology of SBA–Al2O3 composite. Aggregates of fibrous Al2O3 were clearly observed in the image, while the SBA-15 particles were hardly seen. We envisage that Al2O3 fibers were homogeneously dispersed on the external surface of SBA-15 during the preparation. The fibrous structure of AlOOH likely retained in the subsequent calcination step. Cheng et al. reported that the Al2O3 colloid particles could deposit on the surface of SiO2 microspheres via electrostatic attraction strategy at the pH value between the isoelectric points of AlOOH and SiO2.21 It should be emphasized that the pH value of the synthesis solution (pH = 4–5) was appropriate in preparing microspheric SiO2@Al2O3 composites since the isoelectric points of AlOOH and SBA-15 are at pH values of 9.1 and 2.3, respectively.27,28
 | ||
| Fig. 2 SEM images of the as-synthesized SBA-15 (a), AlOOH (b), SBA–Al2O3 (c), and the schematic representation of SBA–Al2O3 synthesis (d). | ||
Fig. 3 displays the N2 adsorption/desorption isotherms and pore size distribution curves of the as-synthesized SBA-15, Al2O3 and SBA–Al2O3 samples. As shown in Fig. 3a, the SBA-15 displayed a typical IV isotherm with two sharp inflections in the relative pressure range of 0.70–0.80 and an H1-type hysteresis loop. The BET surface area and pore size of the parent SBA-15 sample are 906 m2 g−1 and 7.5 nm (Table 1, 1), respectively. A significant quantity of microporous surface area of SBA-15 sample was found (321 cm2 g−1) which is attributed to the microporous corona in the mesopores walls. Fig. 3b displays the characteristic type IV isotherm for the Al2O3 sample, where the adsorbed amount continuously increased with the increase of P/P0, corresponding to the BET surface area of 225 m2 g−1 and the pore size of 9.8 nm (Table 1, 2). The SBA–Al2O3 sample (Fig. 3c) displayed a typical IV isotherm, relevant to the BET surface area of 425 m2 g−1, total pore volume of 0.97 cm3 g−1 and the pore size of 8.9 nm (Table 1, 3). The microporous surface area of the SBA–Al2O3 sample was greatly diminished compared with that of the SBA-15 sample.
 | ||
| Fig. 3 N2 adsorption/desorption isotherms and pore size distribution curves (inset) of the catalysts before (black, top) and after (blue, bottom) steaming at 800 °C for 4 h: SBA-15 (a), Al2O3 (b), SBA–Al2O3 (c), comparison sample SBA/Al2O3-imp (d). | ||
| No. | Samples | Area (m2 g−1) | Volume (cm3 g−1) | BJH diameter (nm) | ||||
|---|---|---|---|---|---|---|---|---|
| Smicro | Sexternal | SBET | Vmicro | Vexternal | Vtotal | |||
| Fresh catalyst | ||||||||
| 1 | SBA-15 | 321 | 585 | 906 | 0.13 | 0.99 | 1.12 | 7.4 |
| 2 | Al2O3 | 0 | 225 | 225 | 0.00 | 0.41 | 0.41 | 9.8 |
| 3 | SBA–Al2O3 | 42 | 383 | 425 | 0.01 | 0.96 | 0.97 | 8.9 |
| 4 | SBA/Al2O3-imp | 84 | 162 | 246 | 0.03 | 0.24 | 0.27 | 6.5 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||||
| Steamed at 800 °C for 4 h | ||||||||
| 5 | Steamed Al2O3 | 0 | 100 | 100 | 0.00 | 0.61 | 0.61 | 33.0 |
| 6 | Steamed SBA–Al2O3 | 4 | 146 | 150 | 0.00 | 0.60 | 0.60 | 18.7 |
| 7 | Steamed SBA/Al2O3-imp | 12 | 32 | 44 | 0.01 | 0.19 | 0.20 | 23.7 |
We also investigated the porous structure of aluminated SBA-15 prepared by conventional impregnation method (denoted as SBA/Al2O3-imp). The Si/Al ratio of 1.04 was chosen for SBA/Al2O3-imp, and the value was similar to that (1.12) of SBA–Al2O3. The SBA/Al2O3-imp (Fig. 3c) showed one-step adsorption isotherm and step-wise desorption isotherm. The step-wise desorption isotherm is due to the fact that the encapsulated mesopores empty at lower pressure than the open pores of similar size.29 The BET surface area of the SBA/Al2O3-imp was 246 m2 g−1.
The hydrothermal stability of silica–alumina composites is of vital importance in practical application. We therefore hydrothermally treated the above-mentioned materials with 100% steam at 800 °C for 17 h and then tested their textural properties. The surface areas of steamed Al2O3, SBA–Al2O3 and SBA/Al2O3-imp decreased to 100, 150 and 44 m2 g−1, respectively. This result suggests that the porous structure of SBA–Al2O3 was more stable than SBA/Al2O3-imp in steam. Early study proposed that Al2O3–SiO2 composites containing crystallized alumina showed higher hydrothermal stability, which well explained our finding that SBA–Al2O3 showed excellent hydrothermal stability. By contrast the surface area of SBA/Al2O3-imp decreased sharply since amorphous Al2O3 was present (ESI, Fig. S1†). The XRD patterns of the steamed catalysts were also shown in Fig. S1.† The steamed fibrous alumina sample transferred to δ-alumina (JCPDS card number 23-1009), while the crystalline structure of γ-alumina phase in the SBA–Al2O3 was retained.
To further characterize the evolution of silicon and aluminum species in the synthesis, these samples were subjected to 27Al MAS NMR analysis and Fig. 4 displays the spectra of the as-synthesized samples. The NMR of AlOOH sample (Fig. 4a) shows a single peak at ∼6 ppm, which attributes to the octahedral coordinated aluminum in the well crystallized boehmite.30 After calcination, the boehmite phase (AlOOH) turned to Al2O3 phase. Accordingly, peak at ∼65 ppm was noticed (Fig. 4b) assigned to tetrahedral coordinated aluminum atoms in clusters of aluminum oxide. Fig. 4c displays the NMR of SBA–AlOOH and two peaks at ∼54 and ∼6 ppm are found. The latter one is due to the octahedral coordinated aluminum of AlOOH, while the former one is attributed to the tetrahedral coordinated aluminum in the vicinity of terminal silanols.16,31 These tetrahedral coordinated Al sites generally show weak Bronsted acidity and have found wide applications in catalysis.32 According to Fig. 4c and d, the ratio of tetrahedral Al species in the vicinity of terminal silanols is estimated to be about 2% of the total Al atoms, and it increases to about 5% upon calcination.
 | ||
| Fig. 4 27Al MAS NMR spectra of the as synthesized AlOOH (a), Al2O3 (b), SBA–AlOOH (c), SBA–Al2O3 (d). The dash lines represent the deconvoluted resonances. | ||
Fig. 5 displays the OH stretching vibrations between 3200 cm−1 and 3800 cm−1 for the SBA-15, Al2O3 and SBA–Al2O3 samples. As shown in Fig. 5a, the SBA-15 showed two bands at ca. 3745 and 3679 cm−1, both corresponding to the stretching mode of Si–OH groups which are free of hydrogen bonding.33 Three characteristic absorption bands of OH groups of Al2O3 were observed at 3720, 3671, and 3586 cm−1 (Fig. 5b).34 Fig. 5c shows the OH vibration of SBA–Al2O3 sample and only bands at 3745 and 3679 cm−1 assigned to Si–OH were noticed, and those peaks corresponded to alumina were hardly seen. For comparison, the IR spectroscopy of the physical mixture of SBA-15 and Al2O3 is also present (Fig. 5d), which clearly shows the vibration bands due to Al–OH. The IR results suggest that the reaction between surface Si–OH in SiO2 and Al–OH species in AlOOH occurred in the preparation, likely leading to the presence of Si–OH–Al sites. It should be noted that the acid strength of thus formed Si–OH–Al species (3745 cm−1) differs from that of bridge OH species in zeolites.
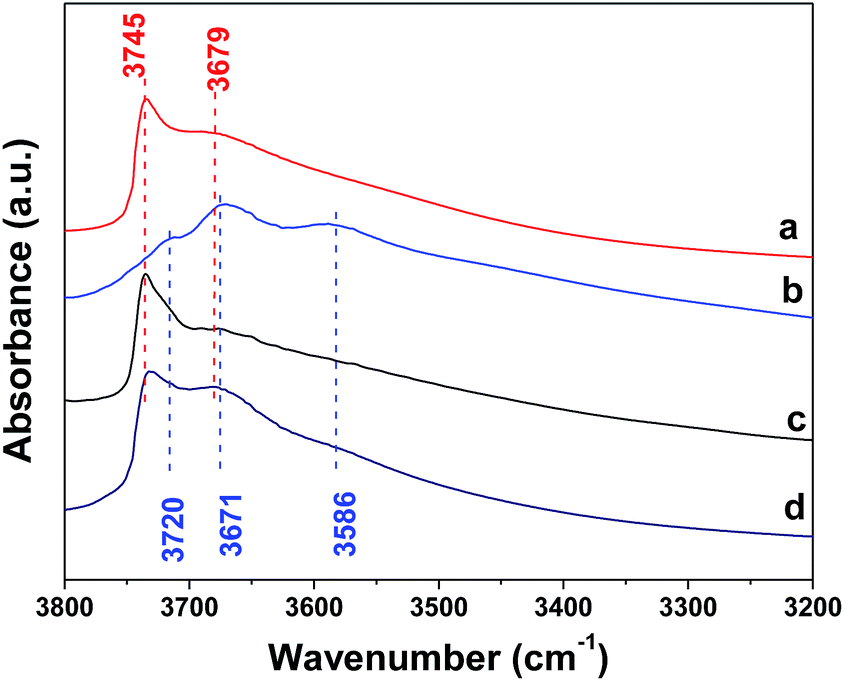 | ||
| Fig. 5 IR spectra (OH region) of the SBA-15 (a), Al2O3 (b), SBA–Al2O3 (c), and physical mixture of 43 wt% Al2O3 and 57 wt% SBA-15 (d). | ||
The acidic properties of these materials were further studied by both pyridine adsorption and NH3-TPD. Fig. 6 displays IR spectra of pyridine absorbed on the catalysts after desorption at 150 °C. The Al2O3 displayed a band at 1450 cm−1 ascribed to pyridine adsorbed on Lewis acid sites.35,36 Pyridine adsorbed on SBA–Al2O3 sample showed two bands at 1544 and 1453 cm−1 related to Bronsted sites and Lewis sites, respectively. The ratio of Bronsted acid sites to Lewis acid sites (B/L) for SBA–Al2O3 sample was lower than that for SBA/Al2O3-imp sample. This result is consistent with the NMR finding that limited amount of tetrahedral coordinated aluminum in the vicinity of terminal silanols were formed, as shown in Fig. 4d. After steaming, the ratio of B/L for the SBA–Al2O3 remarkably increased and surpassed that of SBA/Al2O3-imp catalyst. The total amounts of acid site on these samples were determined by NH3-TPD (see Table 2 and Fig. S2†). The acid site density of SBA–Al2O3 was 0.53 m2 g −1, much higher than that of Al2O3 and SBA/Al2O3-imp sample, in accordance with the higher surface area. After steaming at 800 °C for 4 h, the SBA–Al2O3 sample remained 26.4% of the total acid sites, but the SBA/Al2O3-imp sample only remained 18.8% of the total acid sites.
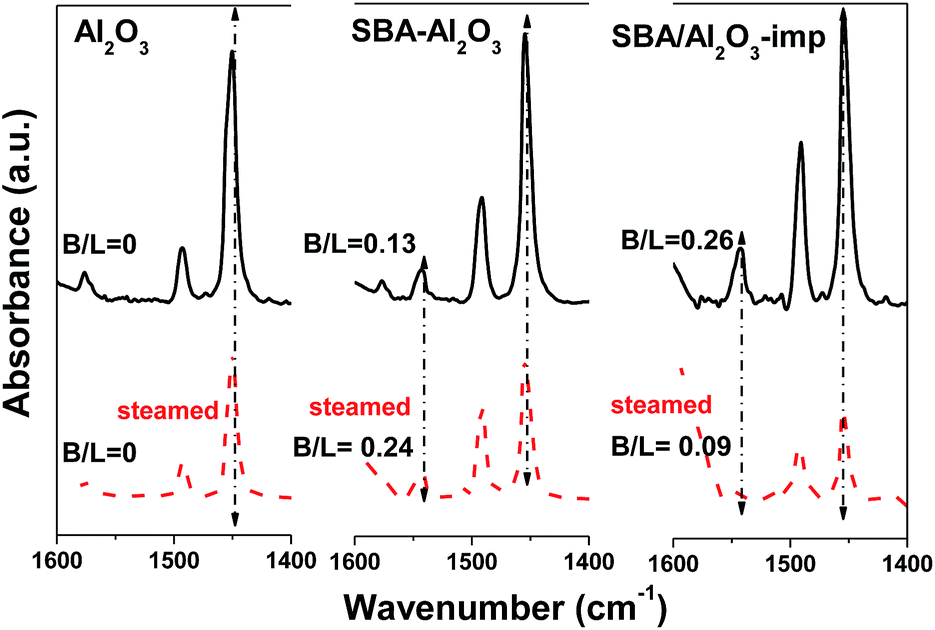 | ||
| Fig. 6 IR spectra of pyridine absorbed on Al2O3, SBA–Al2O3, and reference SBA/Al2O3-imp before (solid) and after (dash) steaming at 800 °C for 4 h (B/L represents the molar ratio of Bronsted acid sites to Lewis acid sites. The absorption coefficients for Bronsted acid sites and Lewis acid sites are 3.03 and 3.80, respectively). | ||
| No. | Catalyst name | Acid sitesa (mmol g−1) | Conv.b (%) | Product yield,b wt% | |||||
|---|---|---|---|---|---|---|---|---|---|
| Propylene | Benzene | Cumene | DIPB | Other CH | Coke | ||||
| a Determined by NH3-TPD measurement.b Conv. of TiPB is defined as the sum of propylene, benzene, cumene, diisopropylbenzene (DIPB) and other hydrocarbons. DIPB is the sum of 1,2-DIPB, 1,3-DIPB and 1,4-DIPB. The total carbon recovery was >95%. | |||||||||
| Fresh catalyst | |||||||||
| 1 | Al2O3 | 0.48 | 2.7 | 0.5 | 0.0 | 0.1 | 1.9 | 0.1 | 0.1 |
| 2 | SBA–Al2O3 | 0.53 | 57.7 | 12.5 | 0.8 | 6.6 | 32.4 | 3.2 | 2.2 |
| 3 | SBA/Al2O3-imp | 0.32 | 23.7 | 4.3 | 0.6 | 2.6 | 12.8 | 1.5 | 1.9 |
|
|||||||||
| Steamed at 800 °C for 4 h | |||||||||
| 4 | Steamed Al2O3 | 0.11 | 0.3 | 0.1 | 0.0 | 0.0 | 0.2 | 0.0 | 0.0 |
| 5 | Steamed SBA–Al2O3 | 0.14 | 31.5 | 6.6 | 0.4 | 1.9 | 20.0 | 1.4 | 1.2 |
| 6 | Steamed SBA/Al2O3-imp | 0.06 | 8.7 | 1.6 | 0.0 | 0.3 | 6.1 | 0.3 | 0.4 |
EtOH-TPD has been shown to be a probe reaction in characterizing the reactivity of surface acid sites (OH groups) on aluminum based oxides by monitoring desorption of ethanol and ethylene. SBA-15 was almost inert in ethanol dehydration to ethylene at temperature below 200 °C (Fig. 7a).37,38 The Al2O3 sample (Fig. 7b) exhibited two desorption peaks at ∼85 and ∼260 °C. The former one corresponds to desorption of medium-strongly bonding ethanol, and the latter one is ascribed to the desorption of formed ethylene on the Al–OH of alumina.38–41 The ethylene desorption peak on SBA–Al2O3 shifts to higher temperature of ∼280 °C as compared to that on Al2O3, as demonstrated in Fig. 7c.
 | ||
| Fig. 7 EtOH-TPD profiles of SBA-15 (a), Al2O3 (b), and SBA–Al2O3 (c). | ||
The main target of this study is to synthesize a binary SiO2–Al2O3 composite with large surface area, improved hydrothermal stability and suitable acidity in a simple way. To this end, high surface area SBA-15 and fibrous AlOOH materials were chosen as the Si and Al precursors, respectively. The resulted SBA–Al2O3 showed surface area of 425 m2 g−1 and total pore volume of 0.97 cm3 g−1, which are both larger than commonly seen binary oxides. SEM results suggest that the morphologies of SBA-15 and AlOOH did not change during the synthesis, while boehmite phase AlOOH turned to crystalline γ-Al2O3, which is thought to stabilize the SBA-15 structure from collapse upon calcination. Moreover, the fibrous morphology may also contribute to the hydrothermal stability of the resulted material based on the finding that least loss of surface area was observed for SBA–Al2O3 after steaming at 800 °C. Another important issue of binary Si–Al oxide synthesis is the introduction of acid sites, which was also achieved by this simply method. It is well known that migration of silicon and aluminum species into each other leads to the formation of weak Si–OH–Al sites.15,42 This process is unambiguously observed by the above mentioned characterization techniques. First the OH vibration due to Al–OH disappeared after being coated on SBA-15. Second, newly formed tetrahedral coordinated aluminum (2%) in the SBA–AlOOH sample was found and its content further increased to 5% upon calcination according to 27Al-NMR. This recombination of Si and Al atoms undoubtedly leads to the formation of weak Bronsted acid sites as well as increased acid density according to pyridine adsorption and NH3-TPD. Last, the higher ethylene desorption peak observed in ethanol desorption tests also pointed to the formation of different acid sites on SBA–Al2O3 from that of pristine Al2O3 phase. Caillot et al. found that desorption peak of ethylene shifted to higher temperature when silica was grafted on the Al–OH of alumina surface due to the loss of Lewis Al3+ sites while forming Bronsted acid sites.38 These results confirm that weak Bronsted sites preferably formed by transfer of Si species from the SBA-15 to the AlOOH or Al2O3 surface, therefore resulting in an acidic binary SBA–Al2O3 material with high surface area and large mesopore.
Catalytic performance
Cracking is the process whereby bulk organic molecules are broken down into smaller molecules such as light hydrocarbons. Zeolite, with high microspore surface area and high acidity, serves as the main active component of FCC catalyst. But some of the bulky hydrocarbons in heavy residues are larger in size than the opening of the microspores of zeolite, which need pre-cracking on the external surface of the zeolite and/or matrix.2 To explore the potential application of thus prepared SBA–Al2O3 as a matrix in FCC, cracking of TIPB with kinetic diameter of 9.4 Å was carried out.Table 2 lists the activities of TIPB cracking on various materials. Al2O3 exhibited a TIPB conversion of 2.7% (Tables 2, 1). By contrast, the SBA–Al2O3 sample gave the TIPB conversion of 57.7% (Tables 2, 2). As expected, propylene, benzene, cumene, and diisopropyl benzene (DIPB) were the most prominent products. The by-products included light hydrocarbons, aromatics and cokes.38,43 The comparison SBA/Al2O3-imp sample exhibited the TIPB conversion of 23.7% (Table 2, 2–3). Generally, the cracking activity are controlled by the acidity and accessibility of the catalysts.44–46 Only weak Lewis acid sites are present on Al2O3 therefore very low activity was observed for Al2O3.38 SBA–Al2O3 catalyst showed higher TIPB conversion than the SBA/Al2O3-imp, because of its higher amount of total acid sites and more open mesoporous channel. Early studies have also shown that introducing mesopores into the zeolites, i.e. ZSM-5, MCM-22, remarkably enhances their catalytic activities in TIPB cracking.47,48 It is believed that the effective diffusion length of bulky molecules in large pores or hierarchical zeolites can be obviously shortened, thus allowing for the improved utilization of acid sites.49 On the other hand, this structural property also led to considerably higher coke yield for SBA–Al2O3 (Fig. S3†). After steaming at 800 °C for 4 h, the initial catalytic activities of SBA–Al2O3 and SBA/Al2O3-imp were 31.5% and 8.7%, respectively (Table 2, 5–6). The loss in TIPB conversion is in line with the decrease of surface area after steaming in critical conditions. Compared with SBA/Al2O3-imp, steamed SBA–Al2O3 still showed much higher reactivity for the cracking of TIPB thanks to its strengthened acidity by the steaming treatment. The activity difference of SBA–Al2O3 and SBA/Al2O3-imp after steaming again points to the vital role of hydrothermally stable porous structure and acid sites in cracking. Previous study also found that Al containing hexagonally ordered MCM-41 showed high activity in TIPB cracking, whereas completely lose its activity after steaming at 600 °C for 2 h. By contrast, a hydrothermally stable mesoporous aluminosilicate still showed very strong acidity and high activity in TIPB cracking.46
Conclusions
In this work, the binary SiO2/Al2O3 composite was synthesized by coating fibrous γ-alumina on the external surface of SBA-15. The SiO2/Al2O3 composite had the surface area of 425 m2 g−1 and pore volume of 0.97 m3 g−1, and no obvious blocked mesoporous was found. The crystalline γ-alumina phase in the mixture was proposed to stabilize the mesoporous structure of SBA-15. The un-calcined binary SiO2/Al2O3 composite contained 2% of tetrahedrally coordinated aluminum which increased to 5% after calcination. The binary SBA–Al2O3 composite prepared by this facile means displayed much higher activity than the SBA/Al2O3-imp material prepared by conventional impregnation method in the cracking of bulky TIPB, and the catalytic activity was well retained after 100% steam treatment at 800 °C for 4 h. It was found that both Bronsted acid sites and Lewis acid sites were present in the binary SBA–Al2O3 composite resulted by the migration of Si and Al in the matrix. Moreover, steaming treatment under critical condition further improved the migration of these atoms leading to strengthened acidity, though with some loss in surface area.Acknowledgements
This work is supported by The National Key Technology R&D Program (No. 2012BAE05B02), The National High Technology Research and Development Program of China (No. 2010AA03A403) and National Science Foundation of China (21533002, 20925310).Notes and references
- G. Busca, Chem. Rev., 2007, 107, 5366 CrossRef CAS PubMed.
- J. Chen, Catalytic Cracking Process and Engineering, China Petrochemical Press, Bei Jing, 2004 Search PubMed.
- T. Isoda, T. Nakahara, K. Kusakabe and S. Morooka, Energy Fuels, 1998, 12, 1161 CrossRef CAS.
- A. Ishihara, H. Negura, T. Hashimoto and H. Nasu, Appl. Catal., A, 2010, 388, 68 CrossRef CAS.
- R. Sadeghbeigi, Fluid Catalytic Cracking Handbook., Elsevier Ltd Oxford, 2012, vol. 47, p. 1 Search PubMed.
- A. Corma, J. Cheminf., 1997, 97, 2373 CAS.
- Q. Li, Z. Wu, B. Tu, S. S. Park, C. S. Ha and D. Zhao, Microporous Mesoporous Mater., 2010, 135, 95 CrossRef CAS.
- I. D. Gay and S. Liang, J. Catal., 1976, 44, 306 CrossRef CAS.
- W. Daniell, U. Schubert, R. Glöckler, A. Meyer, K. Noweck and H. Knözinger, Appl. Catal., A, 2000, 196, 247 CrossRef CAS.
- E. J. M. Hensen, D. G. Poduval, P. C. M. M. Magusin, A. E. Coumans and J. A. R. van Veen, J. Catal., 2010, 269, 201 CrossRef CAS.
- S. A. Tabak and S. S. Shih, Use of high pressure to improve product quality and increase cycle length in catalytic lube dewaxing, EP0104807, Mobil Oil, 1990.
- M. L. Occelli, J. T. Hsu and L. G. Galya, J. Mol. Catal., 1985, 33, 371 CrossRef CAS.
- A. Ishihara, K. Kimura, A. Owaki, K. Inui, T. Hashimoto and H. Nasu, Catal. Commun., 2012, 28, 163–167 CrossRef CAS.
- R. Zhuo, CN Patent 102228839 A, 2011.
- G. T. Whiting, F. Meirer, M. M. Mertens, A. J. Bons, B. M. Weiss, P. A. Stevens, E. d. Smit and B. M. Weckhuysen, ChemCatChem, 2015, 7, 1312 CrossRef CAS.
- G. Busca, Chem. Rev., 2007, 107, 5366 CrossRef CAS PubMed.
- M. F. L. Johnson, J. Catal., 1990, 123, 245 CrossRef CAS.
- B. Beguin, E. Garbowski and M. Primet, J. Catal., 1991, 127, 595 CrossRef CAS.
- H. Jin, Z. Ji, Y. Li, M. Liu, J. Yuan, C. Xu and S. Hou, Colloids Surf., A, 2014, 441, 170 CrossRef CAS.
- H. Jin, L. Xu and S. Hou, J. Mater. Process. Technol., 2010, 210, 81 CrossRef CAS.
- B. Cheng, L. Zhao, J. Yu and X. Zhao, Mater. Res. Bull., 2008, 43, 714 CrossRef CAS.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024 CrossRef CAS.
- Y. Deng, Q. Yang, G. Lu and W. Hu, Ceram. Int., 2010, 36, 1773 CrossRef CAS.
- W. C. Geng, L. B. Duan and Q. Y. Zhang, Appl. Mech. Mater., 2012, 249–250, 992 CrossRef.
- A. Bazyari, A. A. Khodadadi, N. Hosseinpour and Y. Mortazavi, Fuel Process. Technol., 2009, 90, 1226 CrossRef CAS.
- A. Corma, M. Triguero and C. J. MartõÂnez, J. Catal., 2001, 197, 151 CrossRef CAS.
- T. He, L. Xiang and S. Zhu, CrystEngComm, 2009, 11, 1338 RSC.
- M. Kokunešoski, J. Gulicovski, B. Matović, M. Logar, S. K. Milonjić and B. Babić, Mater. Chem. Phys., 2010, 124, 1248 CrossRef.
- P. Van Der Voort, P. I. Ravikovitch, A. V. Neimark, M. Benjelloun, E. Van Bavel, K. P. De Jong, B. M. Weckhuysen and E. F. Vansant, Stud. Surf. Sci. Catal., 2002, 141, 45 CrossRef CAS.
- S. C. Shen, Q. Chen, P. S. Chow, G. H. Tan, X. T. Zeng, Z. Wang and R. B. H. Tan, J. Phys. Chem. C, 2007, 111, 700 CAS.
- J. Jian, K. Johanna, W. Wei, S. S. Ray, F. Hans, F. Dieter and H. Michael, Phys. Chem. Chem. Phys., 2005, 7, 3221 RSC.
- Y. Dai, Y. Zhou, Q. Wei, Q. Cui and Z. Qin, J. Fuel Chem. Technol., 2013, 41, 1502 CAS.
- A. Ungureanu, B. Dragoi, V. Hulea, T. Cacciaguerra, D. Meloni, V. Solinas and E. Dumitriu, Microporous Mesoporous Mater., 2012, 163, 51 CrossRef CAS.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, J. Catal., 2004, 226, 54 CrossRef CAS.
- X. Liu, J. Phys. Chem. C, 2008, 112, 5066 CAS.
- K. Góra-Marek, M. Derewiński, P. Sarv and J. Datka, Catal. Today, 2005, 101, 131 CrossRef.
- M. Caillot, A. Chaumonnot, M. Digne and J. A. van Bokhoven, ChemCatChem, 2014, 6, 832 CrossRef CAS.
- M. Caillot, A. Chaumonnot, M. Digne and J. A. van Bokhoven, ChemCatChem, 2013, 5, 3644 CrossRef CAS.
- J. H. Kwak, D. Mei, C. H. F. Peden, R. Rousseau and J. Szanyi, Catal. Lett., 2011, 141, 649 CrossRef CAS.
- S. Roy, G. Mpourmpakis, D. Y. Hong, D. G. Vlachos, A. Bhan and R. J. Gorte, ACS Catal., 2012, 2, 1846 CrossRef CAS.
- M. A. Christiansen, G. Mpourmpakis and D. G. Vlachos, ACS Catal., 2013, 3, 1965 CrossRef CAS.
- X. Liu, J. Phys. Chem. B, 1999, 103, 2647 CrossRef CAS.
- V. G. Komvokis, S. Karakoulia, E. F. Iliopoulou, M. C. Papapetrou, I. A. Vasalos, A. A. Lappas and K. S. Triantafyllidis, Catal. Today, 2012, 196, 42 CrossRef CAS.
- H. Widdecke, J. Klein and U. Haupt, Macromol. Symp., 1986, 4, 145 CrossRef CAS.
- Y. Lu, M. He, J. Song, X. Shu, Y. Lu, M. He, J. Song and X. Shu, Stud. Surf. Sci. Catal., 2001, 134, 209 CrossRef CAS.
- Z. Zhang, Y. Han, L. Zhu, R. Wang, Y. Yu, S. Qiu, D. Zhao and F.-S. Xiao, Angew. Chem., Int. Ed., 2001, 40, 1258 CrossRef CAS.
- J. Ding, M. Wang, L. Peng, N. Xue, Y. Wang and M.-Y. He, Appl. Catal., A, 2015, 503, 147 CrossRef CAS.
- Y.-J. Ji, H. Xu, D.-R. Wang, L. Xu, P. Ji, H. Wu and P. Wu, ACS Catal., 2013, 3, 1892 CrossRef CAS.
- J. Zhou, Z. Liu, Y. Wang, H. Gao, L. Li, W. Yang, Z. Xie and Y. Tang, RSC Adv., 2014, 4, 43752 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra07467g |
| This journal is © The Royal Society of Chemistry 2016 |