
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural features of mildly fractionated lignin carbohydrate complexes (LCC) from spruce†
Nicola
Giummarella
ab,
Liming
Zhang
b,
Gunnar
Henriksson
ab and
Martin
Lawoko
*ab
aWallenberg Wood Science Center, Royal Institute of Technology, KTH, 100 44, Stockholm, Sweden. E-mail: lawoko@kth.se; Tel: +46 08 7908047
bDepartment of Fiber and Polymer Technology, School of Chemistry, Royal Institute of Technology, KTH, 100 44, Stockholm, Sweden
First published on 22nd April 2016
Abstract
A protocol for the quantitative fractionation of lignin carbohydrate complexes (LCC) from wood under mild conditions has been developed. All operations occur at near-neutral pH conditions and low temperatures, in order to preserve the native structure. The protocol also achieved the fractionation of hemicelluloses of relatively high purity enabling for the first time estimates of hemicelluloses fractions not chemically bound to lignin in wood. 2D HSQC NMR was applied to decipher the structure of LCCs and was complemented by thioacidolysis-GC MS techniques. The carbohydrates linked to lignin in LCC are hemicelluloses, mainly arabinoglucuronoxylan (AGX) and galactoglucomannan (GGM). Benzyl ether (BE) and phenyl glycosidic (PG) linkages were detected. Significant structural differences in the lignin part of LCCs are also reported. The novelty of this work is that we report the first quantitative pH neutral protocol for LCC fractionation and detailed chemical analyses unveil important structural differences of relevance to fundamental knowledge in lignin polymerization and wood-based bio-refineries.
Introduction
Wood consists of three main polymer types namely cellulose, hemicellulose and lignin, which are also the most abundant natural polymers and are presently being investigated for various applications e.g. biomaterials, fuels, platform chemicals and polymers on the basis of renewability.1 Processes that can selectively fractionate the three components in high yield are therefore of interest. To achieve an effective fractionation, it is beneficial to gain insight on how these polymers interact with each other in the native state. The question of the extent to which lignin and the carbohydrates are covalently linked is not yet unequivocally solved. Yet, recalcitrance in biomass fractionation has partly been attributed to these covalent networks between lignin and hemicelluloses.2Three main types of lignin carbohydrate bonds are suggested in the literature namely phenyl glycosides, benzyl ethers and benzyl esters.3 The mechanism for the formation of benzyl ethers and benzyl esters was proposed to be nucleophilic addition reactions of the hydroxyl groups present in carbohydrates or uronic acid moieties to an electrophilic site on a quinone methide intermediate of lignin polymerization.4 The mechanism of formation of the phenyl glycoside is less discussed and requires further investigations to justify its presence. 2D NMR spectroscopy has been an effective tool in studying these linkages.5
Still, concerns remain on the methodologies adopted for fractionation of native LCCs for analytical purposes, since a prerequisite is that they are not formed during fractionation. Mild fractionation techniques have been reported although the yields have ranged from low to moderate.6 When quantitative yields have been achieved, the conditions have been harsher; often including steps involving strong alkaline extractions.7 A universal (applicable to all lignocellulosic biomass) protocol for quantitative fractionation was recently published; however a strong base was applied, in which case some sensitive ester linkages may have cleaved.8 Furthermore, other base catalysed reactions cannot be disregarded.9
In general, LCC fractionation involves sequential dissolution of ball milled wood, followed by selective precipitations normally achieved by the use of anti-solvents or salt addition,10 which also lacks the resolution required to obtain a single molecular species. For this reason, distinguishing LCC from pure lignin or pure carbohydrates in a given fraction by simple compositional analysis cannot be achieved. Detailed structural analysis revealing the connectivity between its compositional polymers is the only evidence of LC bonds.
In the present work, we present a novel technique for the mild quantitative fractionation and characterization of lignin carbohydrate complexes which enabled more reliable and detailed information of the covalent structure of LCC matrix.
Materials and methods
Materials and chemicals
All chemicals used were of analytical grade. Chloroform-d, DMSO-d6, 97% endo-N-hydroxy-5-norbornene-2,3-dicarboximide (e-HNDI), 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane, Amberlite® XAD4 (20–60 mesh) and 1-allyl-3-methylimidazolium chloride were purchased from Sigma Aldrich. Millipore solvent resistant stirred cell (76 mm) for ultrafiltration with 1 kDa regenerated cellulose filter was purchased by Termofisher Scientific (USA). Centrifugation was carried on a Beckman Coulter Avanti J-E equipped with JLA 10.5 and JA 25.5 rotors.Wood mill preparation
Wiley milled wood particles (40 mesh) obtained with a Wiley Mini Mill 3383-L70 from Thomas Scientific, were extracted in a Soxhlet with acetone for 16 h to remove extractives.The extractive free wood meal was dried overnight in a hood and milled according to previous work7 using a Retsch planetary ball mill of type PM400. Two stainless steel jars of 500 ml nominal volume were loaded with 40 g each of sample. Then 100 stainless steel bearings of 10 mm diameter where added to each jar and the milling was performed at room temperature at 300 rpm for 24 hours.
LCCs fractionation
In the first step 50 ml of deionized water was added to 5 g of extractive free ball milled wood and stirred at 80 °C for 4 h. Centrifugation (6300 rpm, 20 min) was done to separate solution 1 from residue 1 (Fig. 1). The solution was passed through polyaromatic resin (Amberlite® XAD4) twice and the permeate (Fraction 1-F1) was separated from the retentate (Fraction 2-F2). The latter fraction was regenerated from the resin with methanol. The resin cycle was repeated three times. The residue after water extraction (residue 1, Fig. 1) was dried overnight in the hood after addition of small amount of acetone and weighed. [Amim]Cl was then added (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 w/w) to it and the mixture obtained was left under stirring at 80 °C for 2 hours. After addition of the same weight of DMSO relative to [Amim]Cl, the solution was kept in same condition for another 2 hours. Finally, 75 ml of DMSO was added and the solution was left overnight under gentle stirring at 70 °C and complete dissolution was achieved. After cooling, deionized water was added (80:20 vDMSO/vwater) and the precipitate (Fraction 3-F3) was separated by centrifugation, washed with and equal volume of water and centrifuged. The supernatant was added to the original solution (solution 2, Fig. 1). The residue was washed again with twice the volume of water and the supernatant discarded. The residue was then freeze dried. To the diluted solution three times its volume of absolute ethanol was added and left to stand overnight at 4 °C. The precipitated (Fraction 4-F4) was separated by centrifugation, dialyzed and freeze dried. Finally, to the remaining solution 3, twice its volume of acetone was added and a precipitate (Fraction 5-F5) was obtained and treated in a similar way to fraction 4.
:1 w/w) to it and the mixture obtained was left under stirring at 80 °C for 2 hours. After addition of the same weight of DMSO relative to [Amim]Cl, the solution was kept in same condition for another 2 hours. Finally, 75 ml of DMSO was added and the solution was left overnight under gentle stirring at 70 °C and complete dissolution was achieved. After cooling, deionized water was added (80:20 vDMSO/vwater) and the precipitate (Fraction 3-F3) was separated by centrifugation, washed with and equal volume of water and centrifuged. The supernatant was added to the original solution (solution 2, Fig. 1). The residue was washed again with twice the volume of water and the supernatant discarded. The residue was then freeze dried. To the diluted solution three times its volume of absolute ethanol was added and left to stand overnight at 4 °C. The precipitated (Fraction 4-F4) was separated by centrifugation, dialyzed and freeze dried. Finally, to the remaining solution 3, twice its volume of acetone was added and a precipitate (Fraction 5-F5) was obtained and treated in a similar way to fraction 4.
 | ||
| Fig. 1 Fractionation scheme for lignin carbohydrate complexes (LCCs). | ||
Carbohydrate analysis
Carbohydrate composition of the lyophilized fractions was determined after acid hydrolysis.11 Briefly, 0.3 ml of 72% sulphuric acid was added to 20 mg of LCCs substrates in a 12 ml glass vial. The mixture was evacuated for 80 minutes in a desiccator, with occasional stirring. After further addition of 8.4 ml of Milli-Q water, the bottle was sealed and left in autoclave for 60 minutes at 125 °C and finally the mixture was filtered by suction pressure through glass fiber filter.Monosaccharide content of the hydrolysate was analyzed with high-performance anion-exchange chromatography (Dionex, Sunnyvale, CA, USA) equipped with pulsed amperometric detector (HPAEC-PAD) using CarboPac PA-1 column (4 × 250 mm) according to previous work.12
The system was equilibrated for 7 minutes with 260 mM NaOH and 170 mM sodium acetate followed by Milli-Q water for 6 min. Only Milli-Q water was used as eluent at a flow rate of 1 ml min−1. A solution of 300 mM NaOH was added to the column effluent before the PAD cell at a flow rate of 0.5 ml min−1. Data was processed with Chromeleon 7.1 software. Anhydro correction of 0.90 and 0.88 were used for hexoses and pentoses, respectively.
Lignin quantitative analysis
Acetobromination was carried on lyophilized fractions according to literature13 where 5 mg of lyophilized fractions were dissolved in 5 ml of glacial acetic acid containing 25% (w/w) of acetyl bromide and 0.1 ml perchloric acid.Then the capped solution was incubated for 30 min at 70 °C with gentle stirring. The vials were placed to cool in an ice bath containing a small amount of ethanol. To the cooled solutions, 5 ml of 2 M NaOH tempered to 4 °C and 12.5 ml glacial acetic acid were added.
The quantification was done on a Shimadzu UV2550 UV/VIS spectrophotometer at 280 nm. The extinction coefficient of the acetobrominated lignin was determined to 25.7 using the known lignin content of spruce of 27% according to previous work.7 Quantification of dioxane soluble lignin was done by dissolving 2 mg of lyophilized fractions in 2 ml of 96% aqueous dioxane for 48 hours at room temperature under continuous and gentle stirring.6 The lignin content was determined using the absorbance values at 280 nm and the extinction coefficient of 19.5, determined from the Klason lignin of the fraction and was in agreement with the literature for spruce milled wood lignin dissolved in dioxane.14
Thioacidolysis and RANEY® desulphuration
To about 20 mg of lyophilized LCC fractions, placed in a vial, a solution of 2 ml of a 2.5% BF3 diethyl etherate with 10% ethanthiol solution in dioxane was added. Then, air was evacuated with nitrogen and the vials capped.15 The samples were heated at 100 °C for 4 hours with gentle shaking every hour. Afterwards, the reaction was stopped by placing the vials in ice bath to cool and then neutralized with 1.5 ml of 0.4 M Na2CO3. Then, about 10 ml of deionized water was added, followed by 5 ml of ethyl acetate and the mixture was vortexed. The organic upper phase consisting of ethylacetate and lignin degradation products was collected. The ethyl acetate extraction was repeated 3 times and then evaporated to dryness under reduced pressure. Desulphuration of thioacidolysis products was done by adding 2 ml RANEY® in aqueous slurry and 5 ml dioxane in a glass tube fitted with a Teflon-lined screw cap and stirring at 80 °C for 4 hours.15 The ice-cooled tube was then cautiously opened and 10 ml of water was added. After adjusting the pH to 3–4, ethyl acetate was added, the mixture vortexed and allowed to form 2 phases (10 minutes). The upper organic phase contained the products. The extraction-phase formation cycle was repeated 3 times. Subsequently the ethyl acetate was removed under reduced pressure to obtain the dried desulphurated samples.Gas chromatography
Gas chromatography with flame ionization detection was performed using Agilent 7890B Gas Chromatograph instrument equipped with a split/splitless injector. The column used was Agilent J&W HP-5 (30 m, 320 μm I.D., 0.25 μm thickness). The content of 4-O-methyl-D-glucuronic acid was determined according to literature16 using erythritol as internal standard. The procedure involves methanolysis, where 5 mg of LCCs were heated at 80 °C for 16 h in 1.2 ml of 2.6 M HCl/MeOH. After cooling, the samples were concentrated under N2 and acetylated according to Önnerud and Gellerstedt17 with pyridine and acetic anhydride (1:1, v/v) at room temperature for 4 hours.
GC-MS was performed with a TRACE™ 2000 GC with Agilent J&W DB-5MS column (30 m, 320 μm I.D., 0.25 μm thickness) and coupled with a Finnigan Trace MS operated at an impact energy of 70 eV.
Galacturonic acid and glucuronic acid were quantified according to Li et al.16 and using the same detector response factor of 0.659.
The desulphurated products of thioacidolysis were quantified using tetracosane as internal standard subsequent to acetylation as previously described.17 The acetylated products were recovered by evaporation after addition of 10 drops of toluene and cold methanol, which was repeated 3 times in order to remove pyridine. The detector response factor of the monomers and dimers necessary for quantification were determined using appropriate model compounds (ESI3†). The yield of thioacidolysis was based on the reaction performed on guaiacylglycerol-beta-guaiacyl ether model (a non-condensed βO4 model) which gave a 76% monomer yield and was consistent with the literature.15 This was also used in the quantitative determination (ESI3†).
SEC-DMSO/0.5% LiBr
Molecular weight distributions of LCCs fractions was investigated by dissolving 5 mg of lyophilized samples in 2 ml of DMSO + 0.5% LiBr (w/w) solution. After filtration of the samples through 0.45 mm PTFE filters, size exclusion chromatography (SEC) was performed with SEC 1260 Infinity (Polymer Standard Services, Germany).The equipment consisted of an isocratic pump (G1310B), a micro degasser (G1379B) and a standard autosampler (G1329B). The detection system included a UV detector (G1314B) in series with a refractive index detector (G1362A). The mobile phase was DMSO + 0.5% LiBr set to a constant flow rate of 0.5 ml min−1 for a total run time of 65 minutes. The injection volume was 100 μl. The separation system consisted of PSS GRAM Precolumn + 2PSS GRAM 100 analytical columns thermostated at 60 °C and connected in series.
The pullulan standards with nominal masses of 708 kDa, 344 kDa, 47.1 kDa, 21.1 kDa, 9.6 kDa, 6 kDa, 1.08 kDa and 342 Da were used for standard calibration according to previous work.18
Acetylation of LCCs fraction
Roughly 200 mg of LCCs fractions were acetylated at same condition reported by Ralph et al.19 LCCs were dissolved in DMSO/N-methylimidazole (2:1, v/v, 4.5 ml) at room temperature for 5 hours and acetylated by adding acetic anhydride (1.5 ml) and gently stirred overnight at room temperature. Then the solution was poured into distilled water (500 ml) to quench the reaction and the resultant precipitate was recovered by centrifugation, washed with same amount of deionized water and lyophilized.
2D HSQC-NMR analysis
For 2D-NMR, 100 mg of the initial LCCs fractions were dissolved in 750 μl of deuterated DMSO-d6 or CDCl3 in the case of acetylated samples. NMR spectra were recorded at room temperature on a Bruker Avance III HD 400 MHz instrument with a BBFO probe equipped with a Z-gradient coil. Sensitivity improved with gradient (e/a TAPPI). The experiments were carried out with the Bruker pulse program ‘hsqcetgpsi’ and the spectra were acquired with the following parameters: size of FID 1024, pulse 9.2 μm, number of dummy scan 16, spectral width 13 ppm and the relaxation delay of 1.5 seconds. The number of scans was set to 120 which lead to a run time of 14 hours. Data were processed with MestreNova (Mestrelab Research) using 90° shifted square sine-bell apodization window; baseline and phase correction was applied in both directions. The unsubstituted carbon 2 of aromatic groups was used as internal standard for quantification as elsewhere.20 The central DMSO (δC/δH = 39.5/2.5 ppm) and chloroform (δC/δH = 77.3/7.2 ppm) were used as internal reference.Results and discussion
In the present work, we report a novel protocol for the quantitative fractionation of LCC at milder conditions than previously reported in the literature. The obtained LCCs were also studied to gain in-depth structural insight.Estimation of carbohydrate-free lignin in ball milled wood
First, we estimate how much of the lignin in ball milled wood (BMW) was not bound to carbohydrates by extraction with a common lignin solvent, 96% aqueous dioxane which is known to extract pure lignin fractions and some lignin with small amounts of carbohydrates attached to it (LCC).6 Only about 10% of the lignin in ball milled wood was dioxane soluble meaning that the pure lignin extractable from the ball milled wood is below 10%. This approximation assumes that there are no steric hindrances or molar mass restrictions effective during extraction. The yield of the dioxane extracted fraction achieved in the present study is lower than that reported (20%) in the literature for spruce.6 This indicates that the milling energy in the present work is lower since it is documented that the yield of dioxane extractable lignin is proportional to the severity of the ball milling.21Protocol for LCC fractionation
A universal LCC fractionation protocol was recently reported by Du et al.8 However, a recent paper by del Rio et al.,22 reports that deacetylation of hemicelluloses occurred, which would imply that ester LCC linkages may also be cleaved. For these reasons, milder protocols are still required to unveil and quantify native LCC structures. In addition, analyses of LC linkages are compromised by their low frequencies. The removal of unbound carbohydrates would therefore benefit the spectroscopic analyses. In previous work, this has been achieved by enzymatic hydrolyses of the carbohydrates.23,33 However undesired enzyme activities e.g. transglycosidation leading to formation of phenylglycosides as described by Yaku et al.37 need to be controlled for. To address the abovementioned concerns about existing LCC protocols, the present protocol (Fig. 1) has achieved the following:| Mass balance | Lignin content | Sugar analysis by HPAEC/PAD, % | 4-O-Methyl GluA | GalA + GluA | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Dioxane 96% soluble | Totala | |||||||||
| % of wood | % of lignin fraction in soluble fraction | % on wood lignin | Ara ±0.1–0.2 | Gal ±0.1–0.2 | Glu ±0.3–2 | Xyl ±0.1–2.6 | Man ±0.2–1.1 | % on xylan | % on fraction | |
| a The total lignin content was obtained by UV spectroscopy after acetyl bromide procedure. Ara = arabinose, Gal = galactose; Glu = glucose; Xyl = xylose; Man = mannose, GluA = glucuronic acid, GalA = galacturonic acid. | ||||||||||
| BMW | 100 | 11 | 100 | 1.6 | 3.0 | 73.2 | 8.6 | 13.6 | 12 | 4 |
| F1 | 9.5 ± 1 | 7.2 | 3 ± 0.5 | 3.4 | 5.5 | 16.5 | 17.3 | 57.3 | 3.5 | 3.5 |
| F2 | 6 ± 1 | 37 | 4 ± 0.5 | 4.5 | 6.5 | 17.3 | 16.4 | 55.3 | 8 | 8 |
| F3 | 66 ± 2 | 4.1 | 61 ± 2 | 0.9 | 1.6 | 86.8 | 4.9 | 5.8 | 3.5 | 0.3 |
| F4 | 9 ± 2 | 11 | 16 ± 4 | 2.3 | 5.2 | 56.5 | 14.0 | 22.0 | 10 | 2.6 |
| F5 | 2 ± 0.5 | 10.7 | 3 ± 0.5 | 4.3 | 11.7 | 20.8 | 13.2 | 50.0 | 4 | 3.3 |
Hot water has previously been applied to extract hemicelluloses from biomass.25
To further investigate possible linkages between lignin and carbohydrates, size exclusion chromatography (SEC) with a dual UV/RI detection was applied. The co-elution of carbohydrates (detected by RI) with lignin (detected by both detectors) is an indicator of LC linkages. The F2, F4 and F5 samples were completely soluble in the LiBr/DMSO solvent system used for the analysis. Interestingly, the UV and RI traces (ESI2†), showed identical traces; a behavior expected if the lignin was covalently bound to the carbohydrate fraction. The F3 was only partly soluble but the soluble part also showed a co-elution of lignin with the carbohydrates, indicating that they were bound. In fact, when this fraction was acetylated and the solubility improved, still co-elution was observed further substantiating bondage. A residue consisting of cellulose was undissolved. When the fraction was acetylated, full solubility was still not achieved, probably due to hornification of cellulose caused by freeze drying.
However, the dissolved fraction is also likely an LCC on the basis of the co-elution. That lignin was linked to some polysaccharide in the isolated fractions was substantiated by 2D HSQC NMR studies.
Analysis of LC bonds and lignin structure
Several techniques were applied to obtain structural information of the LCCs in the present study. 2D HSQC NMR technique was applied to decipher structural difference in LCC lignin,28 and to obtain information on lignin carbohydrate bonds4b,5 as well as carbohydrates structure.29 The inter-monolignol linkages were assigned according to previous work.5,28 To complement the 2D HSQC studies on lignin structure, thioacidolysis, which is a technique that selectively degrades the βO4 linkages in lignin to obtain mostly monomeric and dimeric products,15 was applied.The ethyl thiolated side chain products can be desulphurated with RANEY® to form reduced side chain structures which are acetylated and analyzed by GC-MS30 to provide quantitative information of the monomeric and dimeric products: the former reflecting the amount of non-condensed etherified linkages in lignin and the latter the condensed units. To permit quantification, tetracosane was used as internal standard and model compounds for monomeric and dimeric products were used to determine the total ion chromatographic (TIC) response factors (ESI3†). Most of the GC MS assignments of the structures (Fig. 2) were done based on the known fragmentation patterns of such acetates and are reported in ESI3.†
 | ||
| Fig. 2 GC MS spectrum and peaks assignment of fraction 5 main monomers (up) and dimers (down) obtained after RANEY® desulphurization of thioacidolysis products. | ||
In general the loss of ketene structure (M = 42) from phenolic acetates and loss of acetic acid (M = 60) as a result of the Mc Lafferty rearrangement31 provides information on the number of phenolic and aliphatic acetates respectively. Subsequent fragmentations may include side chain cleavages (Cγ–Cβ and Cβ–Cα) and loss of methanol from methoxy groups. A set of thioacidolysis-derived monomers (m/z; 194, 208, 206, 266; Fig. 2) resulting from the cleavage of non-condensed βO4 linkages, and several dimeric compounds from the cleavage of condensed βO4 structures (m/z; 414, 400, 372, 358; Fig. 2) are reported. The GC MS analysis includes certain structures which cannot been assigned by the 2D HSQC studies such as 4-O-5 structures and 5–5 structures which are not of dibenzodioxin type (structure 3, Fig. 3). Collectively therefore, these techniques provide a tool box for detailed lignin and LCC structural studies.
 | ||
| Fig. 3 Main lignin (1–9) and lignin–carbohydrate (PG, γ-ester, BE) substructures identified in the 2D HSQC spectra of spruce LCCs. | ||
96% dioxane soluble fraction
We first studied the dioxane lignins isolated from the ball milled wood as previously discussed. The 2D HSQC spectrum is presented in Fig. 4 with different color codes for the different substructures (Fig. 3). From the 2D NMR analysis, the inter-monolignol bonds were evaluated quantitatively, with the signals from the C2 position on the aromatic ring used as representatives for the number of monolignols present in each fraction.20 It is known that the C2 position on the aromatic ring of a phenyl propane unit in native lignin is never substituted and thereby can act as an internal standard for quantification of inter-monolignol units.20 The lignin structure is heterogeneous with respect to the inter-monolignol linkages as shown in Table 2. The quantitative analyses of the inter-unit linkages are also in good agreement with those reported for spruce milled wood lignin in the literature (Table 2). | ||
| Fig. 4 HSQC spectra of dioxane lignin (ball milled lignin) in d6-DMSO; the number in subscript indicates the carbon number either in the aromatic or sugar ring. | ||
| P, % | Zhang et al., % | Capanema et al., % | Sette et al., % | |
|---|---|---|---|---|
| βO4 | 37 | 40–43 | 36–45 | 44.7 |
| β5 | 12 | 10–12 | 9 | 10.6 |
| βO4 (DBO) | 4 | 5 | 7 | 3 |
| ββ | 3 | 3.5 | 3 | 3.2 |
| β1 | 2 | 1 | 2.6 | |
| SD | 2 | 1.2–2 | 2 | 0.2 |
| Coniferyl alcohol | 3 | 2 | 2 | |
| Coniferyl aldehyde | 4 | 3 | 4 | |
| α-Carbonyl | 8 |
The dotted circles in the spectra (Fig. 4) show lignin–carbohydrate (LC) bond signals, also defined in Fig. 3. The assignment of LC bonds was made according to the literature.5
The LC linkage signals are weak in this fraction, in line with previous reports and in support of that the linkages are few relative to the abundant inter-monolignol linkages. Benzyl ethers and phenylglycosides were detected in this fraction. Conversely, the benzyl esters or gamma esters were not detected.
Several studies have shown the presence of gamma esters and not the expected benzyl esters5 and attributed this observation to uronosyl migrations of the linkage from the alfa to gamma position.5c
The βO4 content of the dioxane soluble fraction was lower than that of the two major LCC containing fractions (F3 and F4, Table 3). The lignin also contains end groups consisting of coniferyl alcohol structures, oxidized structures such as coniferyl aldehyde and α-carbonyls structures. The α-carbonyl structures are not coupled to βO4 units, otherwise there would be a signal from Cβ–Hβ at δC/δH 81.3/5.4 ppm.32 This is expected since it is suggested that cleavage of sensitive βO4 bonds in lignin renders the macromolecule smaller and soluble. The cleavage yields carbonyl groups on one fraction and new phenolic hydroxyls on the other.6,33 Studies on the stereoselectivity of βO4 degradation under milling conditions have shown that the erythro is preferentially degraded over the threo isomer34 although only a small amount is actually degraded. The α-carbonyls are thus likely a result of the mechanical cleavage of aryl ether linkages during ball milling, consistent with the literature.35
| Dioxane lignin | F1 | F2 | F3 | F3 Ac | F4 | F4 Ac | ||
|---|---|---|---|---|---|---|---|---|
| a GGM = galactoglucomannan; AGX = arabinoglucuronoxylan; GLC = glucan; ( ) = possibly present. X = detected. | ||||||||
| Solubility in d6-DMSO (at 140 mg mL−1) | Full | Full | Full | Partial | Full | Partial | Full | |
| Main carbohydrates detected in solutiona | GGM, AGX | GGM, AGX | AGX | GLC, GGM | GGM, AGX, GLC | GLC, GGM | ||
| Lignin linkage (relative% of C9 unit) | βO4 | 37 | 28 | 23 | 58 | 52 | 41 | 44 |
| β5 | 12 | 5 | 1 | 12 | 13 | 10 | ||
| ββ | 3 | 2 | 6 | 4 | ||||
| DBO | 4 | 53 | 10 | 6 | ||||
| SD | 2 | 2 | 3 | |||||
| End groups (relative% of C9 unit) | Coniferyl alcohol | 3 | 2 | 5 | ||||
| Coniferyl aldehyde | 4 | 6 | 9 | |||||
| α-Carbonyl structure | 8 | 12 | 47 | |||||
| Lignin–carbohydrate bond detected | Phenyl glycoside | X | X | X | ||||
| Benzyl ether type 1 | X | X | X | X | X | |||
| Benzyl ether type 2 | X | X | X | |||||
| γ ester | (X) | |||||||
The warm water extracts of LCC fractionation protocol
The F1 fraction
This fraction consisted mainly of hemicelluloses and small amounts of lignin (Table 1). It was soluble in the d6-DMSO solvent for the 2D NMR study and the spectrum revealed high concentrations of galactoglucomannan (GGM) and arabinoglucuronoxylan (AGX) (ESI6†). Specific signals from the aromatic carbons of lignin (C2, C5, C6) and methoxy groups were detected. Interestingly, the dominating lignin structure is βO4, and mainly as condensed βO4 in the dibenzodioxin (DBO) substructure. Signals from phenyl glycosides were also detected.Thus, it was likely that the aromatics in this fraction which constituted the permeate had not physically adsorbed to the XAD resin during fractionation as discussed earlier, due to the hydrophilic character of the attached polysaccharide.
This assumption is also supported by that dibenzodioxin structures are hydrophobic and their presence in this water soluble fraction may be explained by linkages to water soluble polysaccharides. The cross peaks from the anomeric carbons (internal and terminal) of the carbohydrates are distinguishable from those of the other carbons. The hydroxyl groups on carbon 2 and carbon 3 in mannose units are also partially acetylated, consistent with the literature on native galactoglucomannans.29 The presence of acetyl groups in the hemicelluloses provides evidence of the mildness of this step. In addition to LCC, this fraction is also likely to contain the bulk of the hemicelluloses which are not linked to lignin since previous studies have isolated hemicelluloses with high purity from hot water extracts of mechanically produced pulps.25
The F2 fraction
This fraction consisted mainly of hemicelluloses and some lignin. The fraction was soluble in the d6-DMSO solvent and the 2D NMR studies showed presence of both lignin and carbohydrate signals, although the hemicelluloses signals dominated (ESI6†). The C2 and C3 hydroxyls of the mannose units were also partially acetylated. Methanolysis GC/MS also showed the enrichment of pectins in this fraction (Table 1). Although not quantified due to low signal intensities, phenyl glycosides and benzyl ethers type 1 (Fig. 3) were detected. The fraction was heterogeneous with respect to lignin structure. The total βO4 content was slightly lower than that of the F1 fraction. The dibenzodioxin (DBO) content was also relatively high when compared to dioxane lignin and the major LCC fraction (F3).[Amim]Cl/DMSO soluble fractions of LCC fractionation protocol
The F3 fraction
Considering the mass balance (Table 1), this fraction contained 68% of the wood lignin, most of the cellulose, about 40% of the AGX and roughly 25% of the GGM. Only 27% of this fraction was soluble in the DMSO-d6 at the concentrations required for the 2D NMR studies. The composition of the dissolved phase consisted almost entirely of lignin and xylan (ESI6†). 40% of the F3 lignin (equivalent of 24% of spruce lignin) was in solution. LC linkages were undetectable probably due to the low solute concentration. The SEC analysis, however, suggested the presence of LC bonds. Interestingly, the βO4 content of the lignin in this fraction was highest.In addition, significant amounts of α carbonyl structures resulting from the mechanical cleavage of βO4, as discussed earlier, were detected. The high levels of α carbonyl structures implies that the βO4 levels in the native structure (before milling) within this fraction are significantly higher than the 58% reported here. The high levels of βO4 substructures present in xylan-linked lignin are consistent with previous reported7 based on thioacidolysis studies. Further work to investigate the reasons for these structural differences is required. Due to the poor solubility of the F3 fraction, acetylation was adopted to improve the solubility levels. Close to complete dissolution was obtained and is manifested in the dominance of signals assigned to glucan28b in the 2D NMR spectrum (Fig. 5). Heterogeneity levels in the lignin structure increased when more of fraction was dissolved, although high levels of βO4 where still observed.
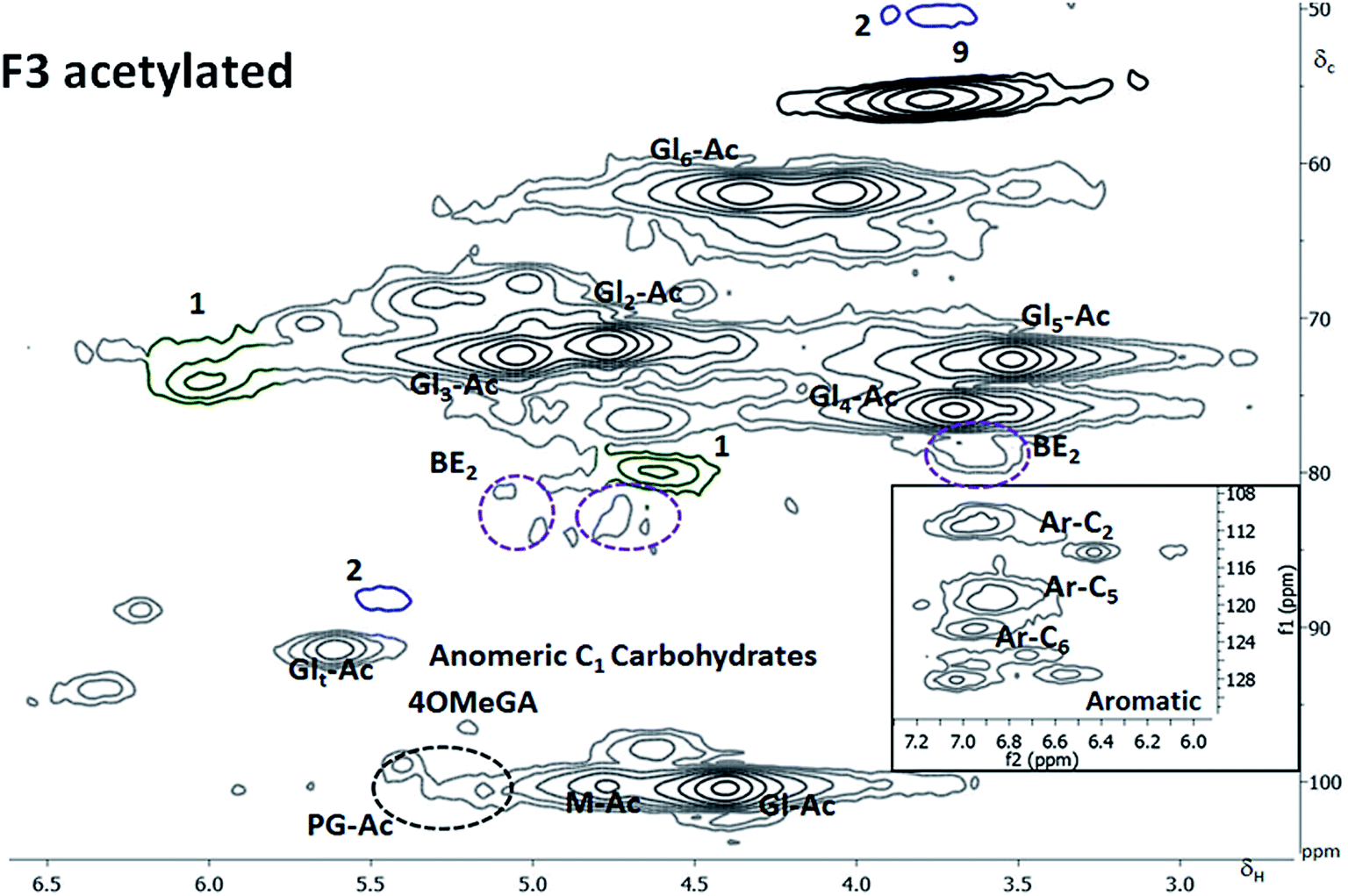 | ||
| Fig. 5 HSQC spectra of fraction 3 after acetylation in CDCl3. M = mannose, Gl = Glucose, Ar-C = carbon in the aromatic, Ac = acetylated, 4OMeGA = 4OMethyl glucuronic acid. The subscript t stands for the carbohydrate terminal reducing end whereas the number in subscript indicates the carbon number either in the aromatic or sugar ring. In the right bottom corner is a zoomed in insert of the aromatic region. | ||
The region where phenylglycosides would appear in acetylated samples was identified by subjecting appropriate model compounds to acetylation followed by 2D NMR analysis (ESI4†). These were also in agreement with work on model compounds recently reported in the literature.5d The analysis showed that about 10% of the inter-unit linkages to lignin in this fraction were phenyl glycosides. This value translates to about 6% of the inter-unit linkages to lignin when the overall mass balance is considered. The phenyl glycosidic bond to mannose C6 unit is the most prevalent. This value is higher than those reported in the literature and is attributed to the mild quantitative fractionation. In previous work, enzymatic hydrolyses have been done to produce cellulolytic enzyme lignins (CEL)5b and enzymatic mild acid lignins (EMAL)36 prior to LCC analysis. The lower levels of phenylglycosides reported in the mentioned work maybe due to that the enzymes, acting on glycosidic bonds in the polysaccharides could also act on phenyl glycosides. There are reports on the cleavage of phenyl glycosidic bonds by β-glucosidases.37 The mechanism of formation of the phenyl glycoside linkage is still not clear from the available literature. One possibility is the acid catalyzed phenolic addition to reducing end of a carbohydrate moiety, by the well known chemistry of acetal formation. If this is the case, then the pH in the cell wall is likely to be slightly acidic when the linkage is formed. Another possibility is the transglycosylation of a phenolic end group to carbohydrate reducing end catalyzed by transglycosidase enzymes. More detailed studies are required to resolve this intrication.
Benzyl ethers were also detected and assigned according to detailed NMR model studies on acetylated samples by Toikka and Brunow.38 The main sugar unit involved in benzyl ether linkage was xylose attached through C2, C3, C4 or C5 to lignin. The cross peaks are reported in the ESI5.† The BE accounted for about 10% of the inter-unit linkages to lignin in the fraction, which, when the total mass balance is considered, is about 6% of the inter-unit linkages to lignin in wood.
Thioacidolysis GC-MS studies of the fraction (Table 4) verified the lignin structures detected by the 2D NMR studies and in addition 4-O-5 linkages not assigned by the 2D NMR studies. Furthermore, the 5–5 structures that are not part of dibenzodioxin structures are included in the GC MS analyses. The major structures revealed by the GC MS were also quantified and are reported in Table 4. Here it should be emphasized that the βO4 studied by the GC MS gives only the non-condensed ethers which is different from that obtained from the 2D HSQC studies. It is evident that the F3 fraction with the highest content of βO4 measured by the 2D HSQC NMR study (Table 3) also had the highest content of non-condensed βO4.
The F4 fraction
This fraction was partially soluble for the 2D NMR analysis. The soluble part was mainly composed of the hemicelluloses and lignin although the whole fraction was mainly cellulose-based. Partial acetylation of the C2 and C3 of the mannose was observed (ESI6†). Again, the preserved acetylation on the galactoglucomannans is testimony to the mildness of fractionation. The βO4 content measured by 2D HSQC NMR was lower than that of the F3 (Table 3) and so was the non-condensed βO4 measured in the GC MS studies (Table 4). The acetylated sample was soluble and the benzyl ether LC bond.(BE 2 type, Fig. 3) was detected and reported in Fig. 6, indicating that xylan was linked to lignin.
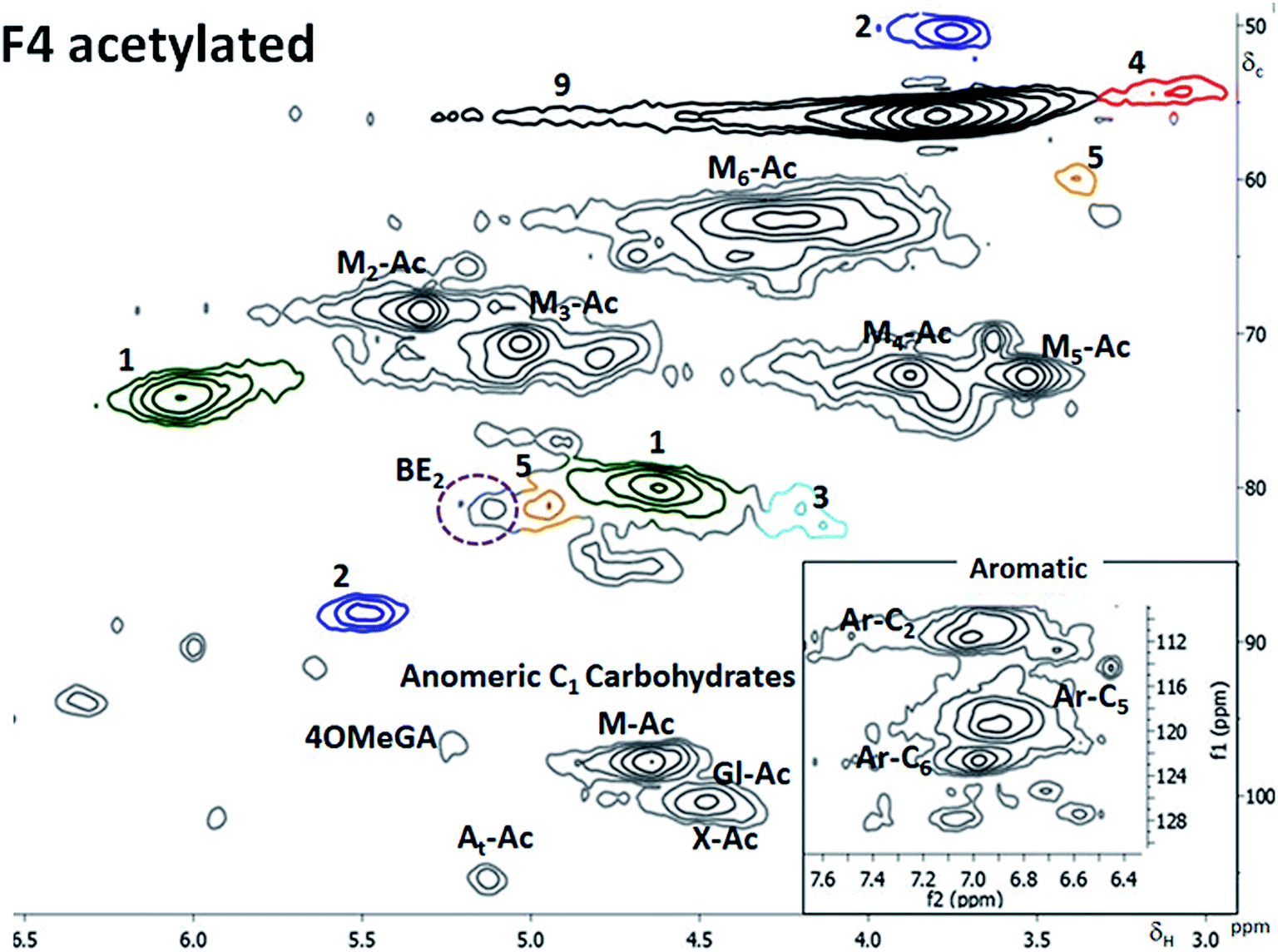 | ||
| Fig. 6 HSQC spectra of fraction 4 after acetylation in CDCl3. In the right bottom corner is a zoomed in insert of the aromatic region. | ||
The signal from BE overlaps with that of spirodienone structure in the non-acetylated LCC; however, when acetylated (Fig. 6), separation is achieved. The BE was linked through C3 in xylose moiety to lignin and constituted 2% of the inter unit linkages in the fraction.
The analytical data shows that LCCs are present in all fractions albeit to different extent. The trend in structural differences between the LCCs for the two analytical methods applied are in agreement. The highest content of βO4 linkages (excluding those involved in dibenzodoxin structure) are found in the F3 fraction, which also had the highest levels of non-condensed βO4 linkages. Herein, both benzyl ethers and phenylglycosides were detected. It is evident, from the overall analytical data, that lignin is mostly linked to the hemicelluloses. The hemicelluloses not bound to lignin, based on our results, would preside in the F1 fraction as reasoned earlier and do not exceed 30% of the total hemicelluloses. The involvement of cellulose in LC linkage was not verified in this study. The LC esters (gamma esters) were unfortunately undetectable due to the region being heavily overlapped.
The structural differences in LCCs lignin present interesting insights into the chemistry of cell wall formation. The xylan linked lignin shows a high degree of homogeneity based on the high proportion of non-condensed βO4. This suggests that the environment in which lignin polymerizes has diverse effects on its structural outcome. On the other hand, the presence of phenylglycoside to mannose in the same fraction could also suggest that this is a xylan–lignin–glucomannan network, i.e. lignin acting as a link between xylan and glucomannan.
Future work should focus on investigating these differences. The structural differences in lignin are also interesting from a polymer science viewpoint. If carefully isolated, homogeneous lignin structures could have better controlled reactivity when subjected to modifications. This could be a good starting point in endeavors to design processes able to fractionate homogeneous lignin subsequently boosting its applicability.
Conclusions
A new mild quantitative method for the fractionation of lignin carbohydrate complexes (LCC) has been developed. The method is universal although only studies of spruce are reported here. Detailed structural studies showed that the main LC linkages in spruce are benzyl ethers (BE) and phenyl glycosides (PG). The main sugar involved in the BE linkage was xylose while mannose was involved in PG linkages. LCC are mainly hemicelluloses-based. Significant structural differences in the lignin part of LCCs were also shown i.e. that lignin covalently bound to xylan was richer in non-condensed βO4 ethers than other lignin.Acknowledgements
This work was supported by Knut and Alice Wallenberg Foundation gratefully acknowledged for financial support to Wallenberg Wood Science Center.References
- (a) A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick Jr, J. P. Hallett, D. J. Leak, C. L. Liotta, R. Jonathan, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311(5760), 484–489 CrossRef CAS PubMed; (b) A. Duval and M. Lawoko, React. Funct. Polym., 2014, 85, 78–96 CrossRef CAS.
- (a) X. Chen, M. Lawoko and A. van Heiningen, Bioresour. Technol., 2010, 101(20), 7812–7819 CrossRef CAS PubMed; (b) M. S. Tunc, M. Lawoko and A. van Heiningen, BioResources, 2010, 5(1), 356–371 CAS.
- D. Fengel and G. Wegener, Lignin–polysaccharide complexes. Wood chemistry, ultrastructure and reactions, ed. W. De Gruyter, Berlin, 1984, pp. 167–174 Search PubMed.
- (a) K. Freudenberg and A. C. Neish, Constitution and biosynthesis of lignin, Springer, Verlag, Heidelberg, 1968, vol. 47, p. 116 Search PubMed; (b) G. Brunow, J. Sipilä and T. Mäkela, Holzforschung, 1989, 43, 55–59 CrossRef CAS.
- (a) M. Y. Balakshin, E. A. Capanema and H.-m. Chang, Holzforschung, 2007, 61, 1–7 CrossRef CAS; (b) M. Y. Balakshin, E. A. Capanema, H. Gracz, H.-m. Chang and H. Jameel, Planta, 2011, 233, 1097–1110 CrossRef CAS PubMed; (c) D. V. Evtuguin, B. J. Goodfellow, C. Pascoal Neto and N. Terashima, Characterization of lignin–carbohydrate linkages in Eucalyptus globulus by 2D/3D NMR spectroscopy using specific carbon-13 labelling technique, Proceedings of the 13th ISWFPC, Auckland, New Zealand. 2005, vol. 2, pp. 439–444 Search PubMed; (d) Y. Miyagawa, T. Mizukami, H. Kamitakahara and T. Takano, Holzforschung, 2014, 68(7), 747–760 CrossRef CAS.
- A. Björkman, Sven. Papperstidn., 1956, 59, 477–485 Search PubMed.
- M. Lawoko, G. Henriksson and G. Gellerstedt, Biomacromolecules, 2005, 6(6), 3467–3473 CrossRef CAS PubMed.
- X. Du, M. Perez-Boada, C. Fernandez, J. Rencoret, J. C. del Rio, J. Jimenez-Barbero, J. Li, A. Gutierrez and A. T. Martinez, Planta, 2014, 239(5), 1079–1090 CAS.
- Ö. Eriksson, D. A. I. Goring and B. O. Lindgren, Wood Sci. Technol., 1980, 14, 267–279 CrossRef.
- M. Lawoko, Int. J. Biol. Macromol., 2013, 62, 705–713 CrossRef CAS.
- SCAN, Carbohydrate composition, 2009, SCAN-CM, 71, 09.
- S. Azhar, G. Henriksson, H. Theliander and M. Lindström, Carbohydr. Polym., 2015, 117, 19 CrossRef CAS PubMed.
- W. Vermerris and R. Nicholson, Phenolic Compound Biochemistry, Springer, 2009, vol. 4, p. 165 Search PubMed.
- A. Rezanowitch, W. Q. Yean and D. A. I. Göring, Sven. Papperstidn., 1963, 66, 141–149 Search PubMed.
- C. Lapierre, B. Pollet, B. Monties and C. Rolando, Holzforschung, 1991, 45, 61–68 CrossRef CAS.
- J. Li, K. Kisara, S. Danielsson, M. E. Lindström and G. Gellerstedt, Carbohydr. Res., 2007, 342(11), 1442–1449 CrossRef CAS PubMed.
- H. Önnerud and G. Gellerstedt, Holzforschung, 2003, 57(2), 165–170 CrossRef.
- A. Duval, F. Vilaplana, C. Crestini and M. Lawoko, Holzforschung, 2015, 69 Search PubMed.
- F. Lu and J. Ralph, Plant J., 2003, 35, 535–544 CrossRef CAS PubMed.
- M. Sette, R. Wechselberger and C. Crestini, Chem.–Eur. J., 2011, 17, 9529–9535 CrossRef CAS PubMed.
- (a) E. Capanema, M. Balakshin, R. Katahira, H.-m. Chang and H. Jameel, J. Wood Chem. Technol., 2014, 35(1), 17–26 CrossRef CAS; (b) Z. Hu, T. F. Yeh, H.-m. Chang, Y. Matsumoto and J. F. Kadla, Holzforschung, 2006, 60, 389–397 Search PubMed.
- J. C. del Rio, P. Prinsen, M. Cadena Edith, A. T. Martınez, A. Gutierrez and J. Rencoret, Planta, 2016, 243(5), 1143–1158 CrossRef CAS PubMed.
- M. Y. Balakshin, E. A. Capanema and A. Berlin, Isolation and Analysis of Lignin-Carbohydrate Complexes (LCC) Preparations with Traditional and Advanced Methods, Studies in Natural Products Chemistry, Elsevier, 2014, vol. 4, pp. 83–115 Search PubMed.
- T. J. Schwartz and M. Lawoko, Bioresources, 2010, 5, 4 Search PubMed.
- S. Willför, K. Sundberg, M. Tenkanen and B. Holmbom, Carbohydr. Polym., 2008, 72, 197–210 CrossRef.
- (a) N. Sun, H. Rodriguez, M. Rahman and R. D. Rogers, Chem. Commun., 2011, 47(5), 1405–1421 RSC; (b) P. Maeki-Arvela, I. Anugwom, P. Virtanen, R. Sjoeholm and J. P. Mikkola, Ind. Crops Prod., 2010, 32(3), 175–201 CrossRef CAS.
- (a) L. Kyllönen, A. Parviainen, D. Somdatta, M. Lawoko, M. Gorlov, I. Kilpeläinen and A. W. T. King, Green Chem., 2013, 15, 2374–2378 RSC; (b) M. T. Clough, K. Geyer, P. A. Hunt, S. Son, U. Vagt and T. Welton, Green Chem., 2015, 17, 231–243 RSC.
- (a) H. Kim, J. Ralph and T. Akiyama, BioEnergy Res., 2008, 1, 56–66 CrossRef; (b) L. Zhang and G. Gellerstedt, Magn. Reson. Chem., 2007, 45, 37–45 CrossRef CAS PubMed; (c) J. Rencoret, G. Marques, A. Gutierrez, L. Nieto, J. I. Santos, J. Jimenez-Barbero, A. T. Martinez and J. C. del Rio, Holzforschung, 2009, 63, 691–698 CrossRef CAS.
- (a) A. Teleman, J. Lundqvist, F. Tjerneld, H. Stålbrand and O. Dahlman, Carbohydr. Res., 2000, 329, 807–815 CrossRef CAS PubMed; (b) A. Teleman, M. Nordstrom, M. Tenkanen, A. Jacobs and O. Dahlman, Carbohydr. Res., 2003, 338(6), 525–534 CrossRef CAS PubMed.
- (a) C. Lapierre, B. Pollet, M. Petit-Conil, G. Toval, J. Romero, G. Pilate, J. C. Leple, W. Boerjan, V. V. Ferret, V. De Nadai and L. Jouanin, Plant Physiol., 1999, 119(1), 153–164 CrossRef CAS PubMed; (b) H. Önnerud, Holzforschung, 2003, 57(4), 377–384 CrossRef.
- D. G. I. Kingston, J. T. Bursey and M. M. Bursey, Chem. Rev., 1974, 74(2), 215–242 CrossRef CAS.
- E. A. Capanema, M. Y. Balakshin, C.-L. Chen, J. S. Gratzl and H. Gracz, Holzforschung, 2001, 55, 302–308 CAS.
- H.-m. Chang, E. B. Cowling, W. Brown, E. Adler and G. Miksche, Holzforschung, 1975, 29, 153–159 CrossRef CAS.
- Z. Wang, T. Yokoyama, H.-m. Chang and Y. Matsumoto, J. Agric. Food Chem., 2009, 57(14), 6167–6170 CrossRef CAS PubMed.
- L. Zhang and G. Gellerstedt, 10th International Symposium on Wood and Pulping Chemistry, 1999, vol. 2, pp. 164–170 Search PubMed.
- A. Guerra, I. Filipponen, L. A. Lucia and D. S. Argyropoulos, J. Agric. Food Chem., 2006, 54, 9696–9705 CrossRef CAS PubMed.
- F. Yaku, Y. Yamada and T. Koshiyima, Holzforschung, 1976, 30, 148–156 CrossRef CAS.
- (a) M. Toikka and G. Brunow, J. Chem. Soc., Perkin Trans. 1, 1999, 1, 1877–1883 RSC; (b) J. Sipilä and G. Brunow, Holzforschung, 1991, 45, 3–7 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: UV measurements, SEC chromatograms, GC-MS defragmentations, 2D HSQC spectra. See DOI: 10.1039/c6ra02399a |
| This journal is © The Royal Society of Chemistry 2016 |