
Esterase-responsive polymeric prodrug-based tumor targeting nanoparticles for improved anti-tumor performance against colon cancer†
Abstract
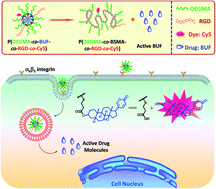
We report on the fabrication of a multifunctional polymeric prodrug covalently linked with an anticancer drug (bufalin, BUF) and tumor-targeting peptide (RGD) and investigate its anticancer performance against colon cancer in mice. The polymerizable monomer, 3-((2-(methacryloyloxy)ethyl) thio)propanoic acid (BSMA), was synthesized first. Atom radical transfer polymerization (ATRP) of BSMA and oligo(ethylene glycol) monomethyl ether methacrylate (OEGMA) afforded random copolymers, P(OEGMA-co-BSMA). The polymeric prodrug of BUF, P(OEGMA-co-BUF), was obtained by an esterification reaction between the carboxyl groups of P(OEGMA-co-BSMA) and the hydroxyl group of BUF. Finally, a tumor-targeting polymeric prodrug, P(OEGMA-co-BUF-co-RGD), was obtained via an aminolysis reaction of P(OEGMA-co-BUF) in the presence of RGD and the final drug content was determined to be ∼32.9 wt%. In aqueous media, P(OEGMA-co-BUF-co-RGD) self-assembles into micelles and the hydrodynamic diameter (Dh) of the micelles was determined to be ∼33.0 (±2.5) nm by dynamic laser light scattering (LLS). It was demonstrated that the tumor-targeting polymeric prodrug showed improved anticancer performance both in vitro and in vivo in comparison with that of free BUF.
 Please wait while we load your content...
Please wait while we load your content...

