
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in the synthesis of benzimidazol(on)es via rearrangements of quinoxalin(on)es
Vakhid A.
Mamedov
*
A. E. Arbuzov Institute of Organic and Physical Chemistry, Kazan Scientific Center of the Russian Academy of Sciences, Arbuzov str. 8, Kazan, 420088, Russian Federation. E-mail: mamedov@iopc.ru
First published on 12th April 2016
Abstract
This is the first review describing all the quinoxaline–benzimidazole rearrangements as a whole and the new quinoxalinone–benzimidazol(on)e rearrangements in particular when exposed to nucleophilic rearrangements for the synthesis of various biheterocyclic motifs. The scope of the rearrangements is illustrated by way of numerous examples of their application, and in doing so, the review contains over 131 references and covers all of the literature, from the first report of the rearrangement of 2,3-diphenylquinoxaline by Ogg and Bergstrom in 1931 up to more recent examples in the past few years. The mechanisms for the selected transformations are also discussed.
 Vakhid A. Mamedov | Vakhid A. Mamedov graduated from the Azerbaijan State University in Baku (1981). After working for few years as research scientist at the Institutes of the Azerbaijan Academy of Sciences he moved (1985) to the A. E. Arbuzov Institute of Organic and Physical Chemistry (IOPC), Kazan Scientific Centre Russian Academy of Sciences, where he obtained his Ph.D. (1989) with Professor Il'dus A. Nuretdinov, and D.Sc. (1999) degrees in Chemistry collaborate with Professors Yakov A. Levin and Eugeniy A. Berdnikov. He spent his postdoctoral fellowship (1997–1998) with Professor Sadao Tsuboi at the Okayama University (Japan), working on synthetic approaches to the C-13 side chain of paclitaxel (Taxol) and docetaxel (Taxotere). He became head of the laboratory of the Chemistry of Heterocyclic Compounds in IOPC in 2002. He authored over 170 papers. Mamedov's research interests focus on organic synthesis methodology and development of new schemes for tandem transformations for synthesizing diversity-oriented heterocyclic systems. |
1 Introduction
Benzimidazole, firstly described by Hobrecker in 1872,1 is an important privileged heterocyclic motif2–5 and one of the most widely investigated scaffolds by synthetic chemists because of its medicinal importance. The benzimidazole scaffold acts as an important class of heterocyclic compounds with a wide range of biological properties.6 Benzimidazole derivatives are structural isosteres of naturally occurring nucleotides, which allow them to easily interact with the biopolymers of the living systems and different kinds of biological activity have been obtained. 2-Aminobenzimidazoles proved useful for acid/base catalysis and can substitute guanidinium groups in receptor molecules designed as phosphoryl transfer catalysts.7 Some 2-aminobenzimidazoles displayed an appreciable antimicrobial effect and their corresponding carbamate derivatives have been synthesized for their significant antifilarial activity in vivo.8 As to their high affinity towards a variety of enzymes and protein receptors, they could be considered as pivotal structures in drug design.9 The optimization of benzimidazole-based structures has resulted in marketed drugs, e.g. omeprazole10 and pimobendan11 which are used as therapeutic agents in the treatment of peptic ulcer and congestive heart failure respectively. Benzimidazole derivatives proved of great interest because of their wide range of biological functions12 and pharmacological applications.13 They are an integral part of various clinical medicines14 as well, for example 2-substituted benzimidazole, esomeprazole15 is an anti-ulcer drug and albendazole16 is used to treat parasitic diseases, whereas, 1,2-substituted benzimidazole, astemizole is an antihistamine drug (Fig. 1).17 Many derivatives of benzimidazoles are well known for their antimicrobial,18 anthelmintic,19 antiviral,20 and antifungal21 activities. The antifungal agent benomyl was first reported as a fungicide against a wide range of agricultural fungal diseases.22 Later on, it proved to be a potent antiproliferative agent against the Hela cancer cell line and could be partly used in cancer chemotherapy.23 Benzimidazole derivatives with ester groups on the benzene ring possessed antifungal, insecticidal and herbicidal activities.24 Besides, many dichlorobenzimidazoles appeared very effective against methicillin resistant Staphylococcus aureus (MRSA).25 Since 1985, benzimidazole containing compounds have been reported as well known anticancer agents.26 The role of mammalian DNA topoisomerases as molecular targets for anticancer drugs received appreciation. Some benzimidazoles have been reported as topoisomerase inhibitors e.g. Hoechst 33258 and Hoechst 33342 (Fig. 1).27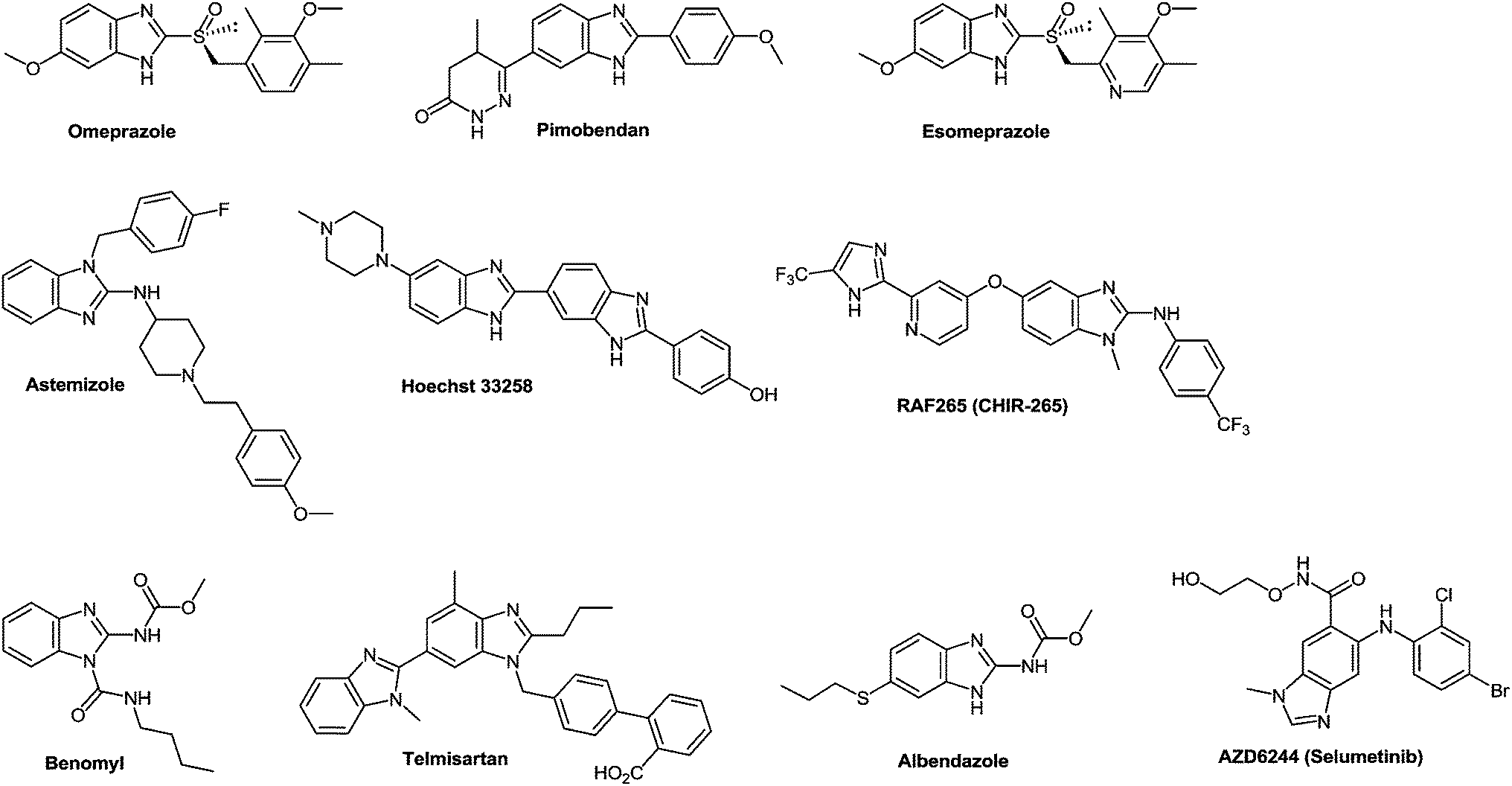 | ||
| Fig. 1 Clinically used benzimidazoles from the current literature. | ||
Some widely used anticancer drugs such as RAF265 (CHIR-265; Novartis Pharmaceuticals, Basel, Switzerland) and AZD6244 (ARRY-142886; AstraZeneca, London, England) are known to contain a benzimidazole moiety. RAF265 resulted in the reduction of tumor cell growth and in tumor cell apoptosis.28 Compound AZD6244 suppresses the growth of melanoma cells through the induction of cytostasis.29 2-Arylbenzimidazole moiety proved to be a pharmacophore for a new class of DNA intercalating agents.30 The importance of naphthalene benzimidazole compounds as antioxidants on hepatic cytochrome has been explored since 1997.31 On the other hand, the antiviral activity of 5-chloro and 5,6-dichloro-2-substituted benzimidazole derivatives against several viruses, e.g. influenza, human cytomegalovirus, hepatitis B virus (HBV), hepatitis C virus (HCV) and human immunodeficiency retrovirus (HIV-1) was reported.32 These compounds proved to be anticancer agents against breast and prostate cancer cell lines33 as well as potential topoisomerase II inhibitors.34 In 2010, a new series of 2-substituted benzimidazole derivatives with a 5-chloro or 5-underivatized carboxylic acid group exhibited antitumor activity against hepatocellular carcinoma (HEPG2), human breast adenocarcinoma (MCF7) and human colon carcinoma (HCT 116) cell lines.35
Telmisartan is a potent angiotensin II receptor antagonist in the treatment of essential hypertension.36 It is one of the most efficient drugs in its class, boasting the longest half-life, a high protein binding affinity, and a low daily dosage.37 The drug is currently marketed under the brand name of Micardis and provides additional benefits against vascular and renal damage caused by diabetes and cardiovascular disease.38
Bis-heterocyclic compounds offer better binding opportunities with a enzyme active site due to its three dimensional special arrangement and consequently possess many intrinsic biological properties.39 In particularly, bis-heterocycles comprising pyridobenzimidazoles (I) are ligands for the BZD site on GABA-A receptors and are therefore used for the treatment of disorders of the central nervous system including convulsion such as epileptic seizures, anxiety, depression, muscular spasm, and attention deficit hyperactivity disorder (Fig. 2).40 Polycyclic bis-heterocycles with imidazopyridine or imidazoisoindole moieties (II) prove the basic structural frameworks of potent inhibitors of respiratory syncytical virus while some imidazoisoindoles (III) exhibit antiviral activity.41 Benzimidazoles in combination with the pyrrolo-isoidolones (IV) exhibit anticancer activity via the inhibition of the ATPase-type catalytic activity of the Hsp90 chaperone protein.42
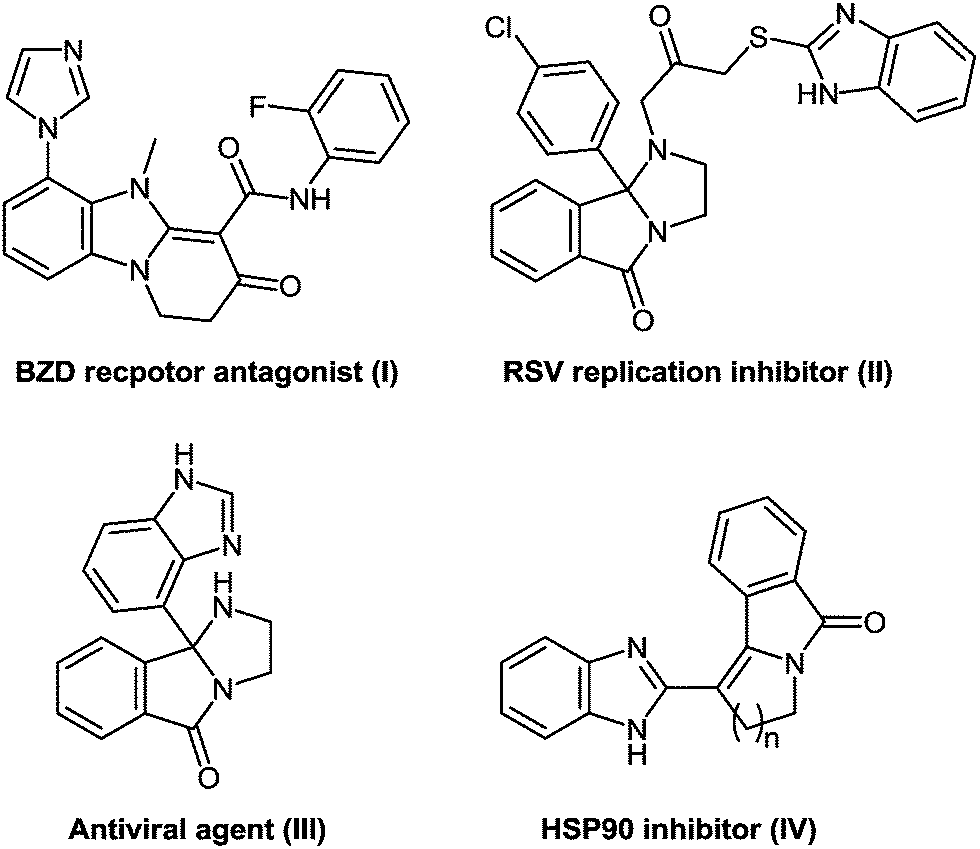 | ||
| Fig. 2 Biologically active bis-heterocyclic compounds. | ||
The drug reference books by Mashkovskiy43 and Negwer44 comprise 92 benzimidazole derivatives used in medicine. However, only 13 of them (Fig. 3) include other heterocyclic rings directly connected with the benzimidazole system. This is apparently due to the fact that such compounds are mostly inaccessible. The analysis of the literary data shows that the methods of the synthesis of such compounds involve many steps, i.e. they are laborious, and the total yields of products never exceed 20% ± 25%.
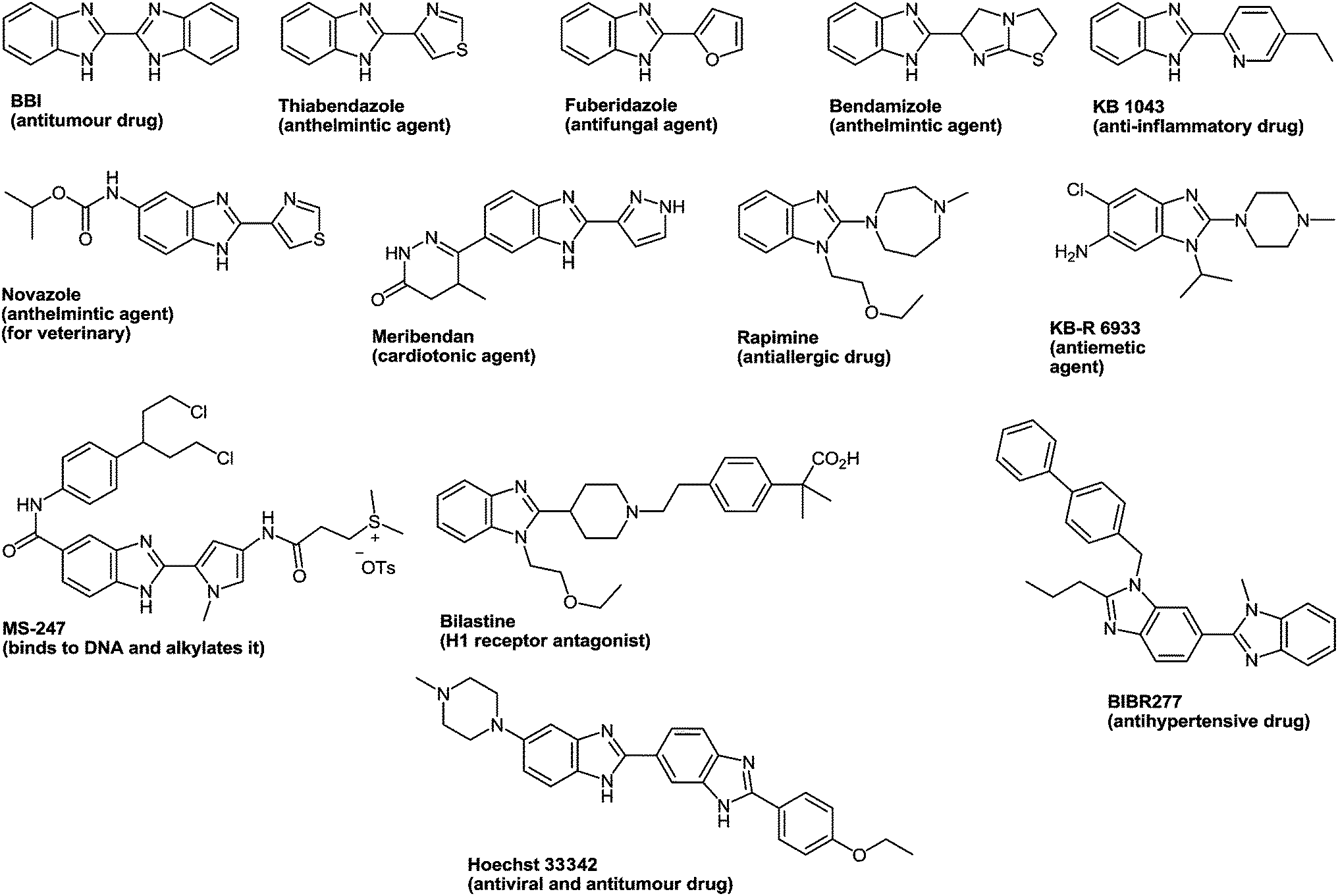 | ||
| Fig. 3 Clinically used hetarylbenzimidazoles from the drug reference books by Mashkovskiy43 and Negwer.44 | ||
There are two classical methods for benzimidazole synthesis, i.e. coupling of 1,2-diaminobenzenes (1,2-DABs) with carboxylic acids and of 1,2-DABs with aldehydes and ketones (the Phillips–Ladenburg and the Weidenhagen reactions, respectively).45 The classical version of these reactions are limited by the use of high temperatures (sometimes, 250–300 °C) and by the low yields of products.46 Actually, all the methods of benzimidazole synthesis which currently exist represent modifications of the reactions mentioned.47
The analysis of the data published has shown that the main drawback of these methods involve the impossibility to use them for synthesizing various types of benzimidazole derivatives. For example, it is no so easy task to enter any given heterocycle in position 2 of benzimidazole ring using of these methods. In addition to the methods mentioned, examples of the formation of benzimidazole derivatives by rearrangement of heterocyclic systems have been documented. Despite the fact that the publications on these reactions are much fewer as compared with the Phillips–Ladenburg and Weidenhagen reactions, they are more diverse but unfortunately not general. The generalization and systematization of the data published on the rearrangement reactions will considerably facilitate the quest of organic chemists for the methods of the synthesis of benzimidazole derivatives inaccessible by the Phillips–Ladenburg and Weidenhagen reactions. In this review, we will focus mainly on recent advances in the synthesis of benzimidazoles and benzimidazolones via new rearrangements of quinoxalinones when exposed to nucleophilic reagents and earlier works of importance will also be discussed.
2 Synthesis of benzimidazoles
2.1 Rearrangement of quinoxalines (historical background)
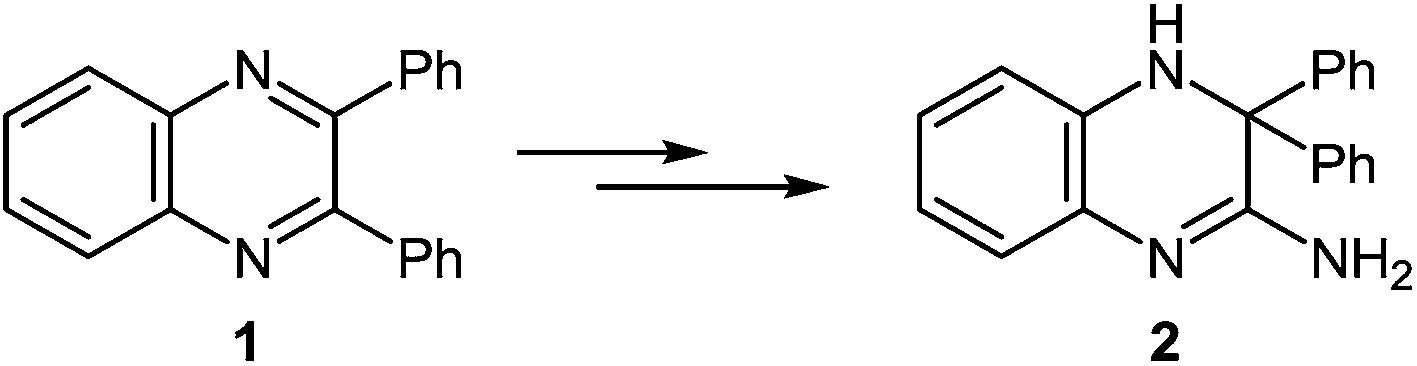 | ||
| Scheme 1 Proposed benzyl → benzilic rearrangement. | ||
The formation of 2-phenylbenzimidazole 3 from 2,3-diphenylquinoxaline 1 and potassium amide involves the initial addition of an amide ion at the C(2) carbon atom, according to Ogg and Bergstrom had postulated, but with subsequent ring contraction, presumably with the elimination of benzylidenimine. This would result in the observed product 3, rather than the phenyl migration of the benzyl acid-rearrangement type (Scheme 2).
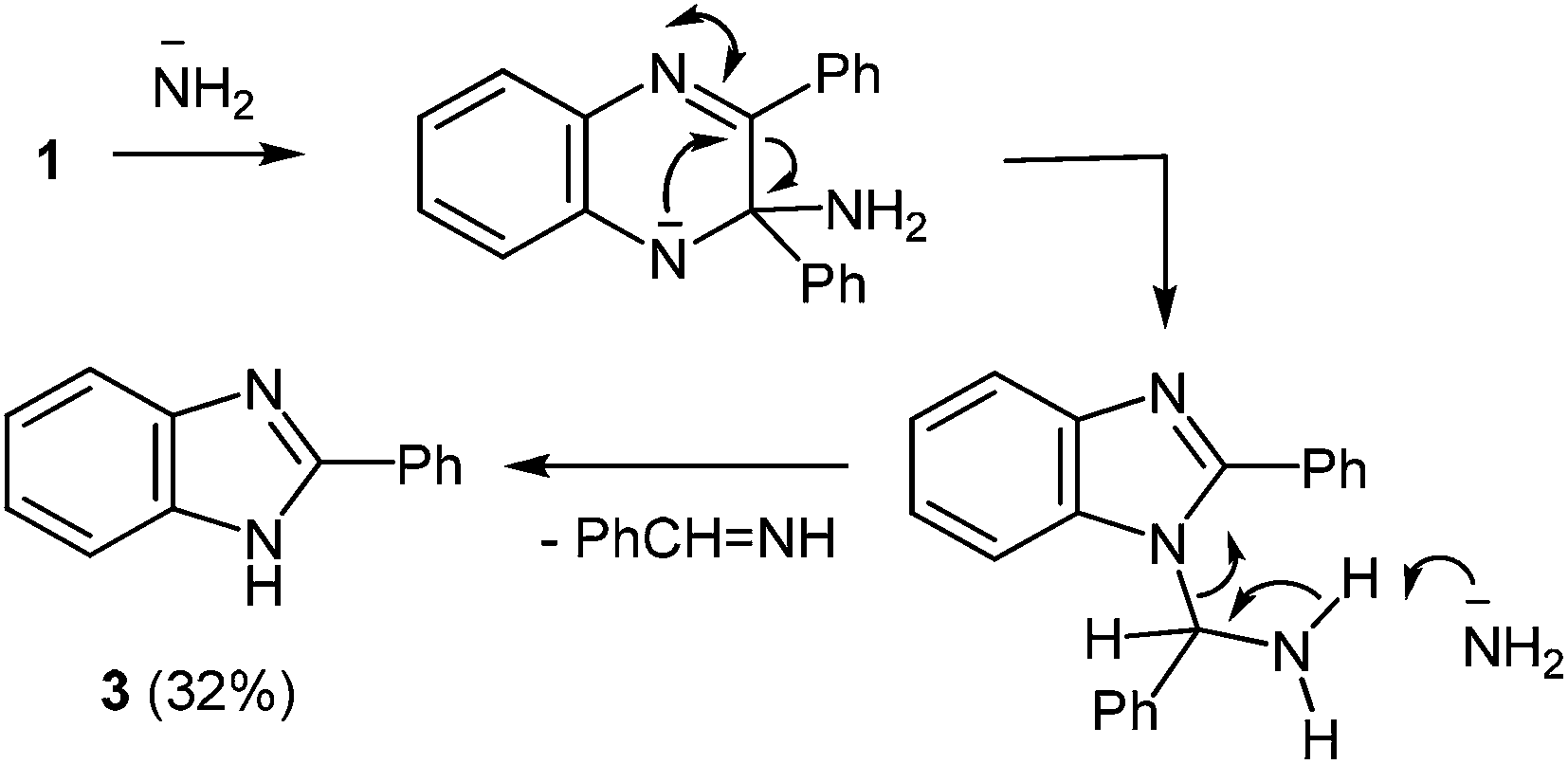 | ||
| Scheme 2 Proposed mechanism of the rearrangement. | ||
Attempts to accomplish the ring contraction of 2,3-diphenylquinoxaline 1 to 2-phenylbenzimidazole 3 with other bases (KOH in H2O or EtOH, NaOH in MeOH, NaH in toluene) were unsuccessful. The efficiency of KNH2 appears to be specific.
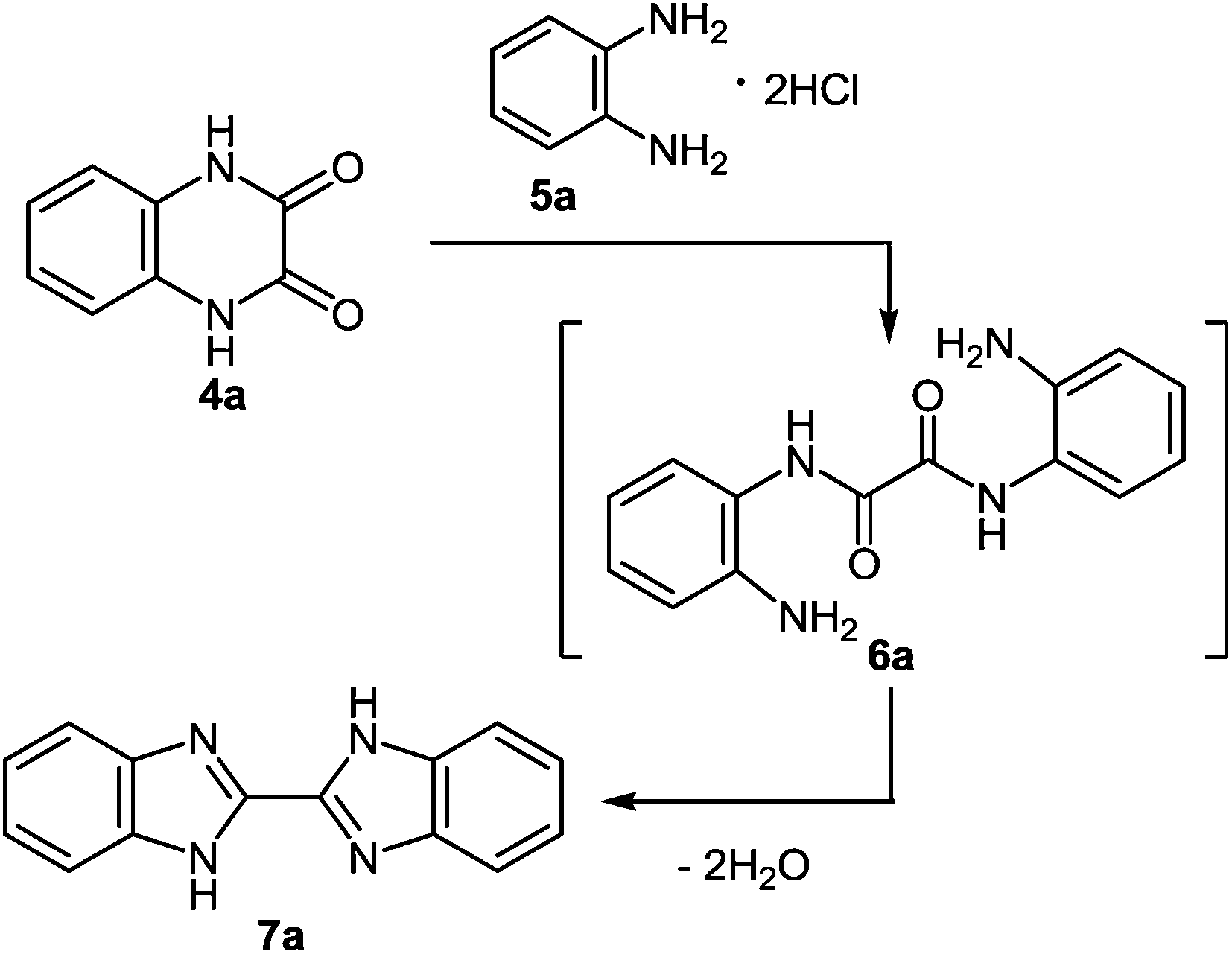 | ||
| Scheme 3 Synthesis of 2,2′-bibenzimidazole. | ||
This reaction sequence has been used for the preparation of an unsymmetrically substituted 2,2′-bibenzimidazole, since 2,3-dihydroxy-6-methylquinoxaline 4b and 1,2-DAB dihydrochloride reacted to form 5-methyl-2,2′-bibenzimidazole 7b (Scheme 4).50
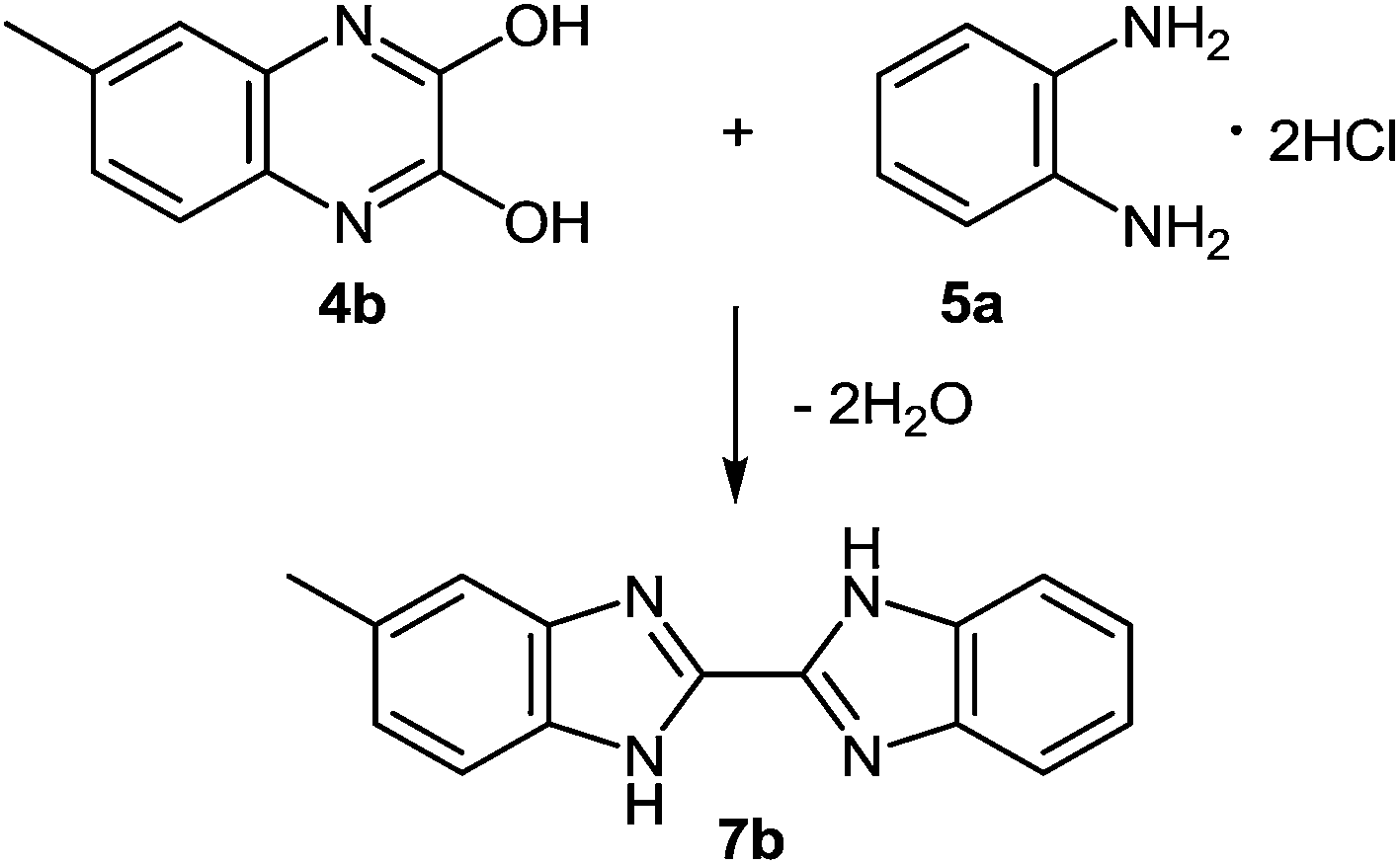 | ||
| Scheme 4 Synthesis of an unsymmetrically substituted 2,2′-bibenzimidazole. | ||
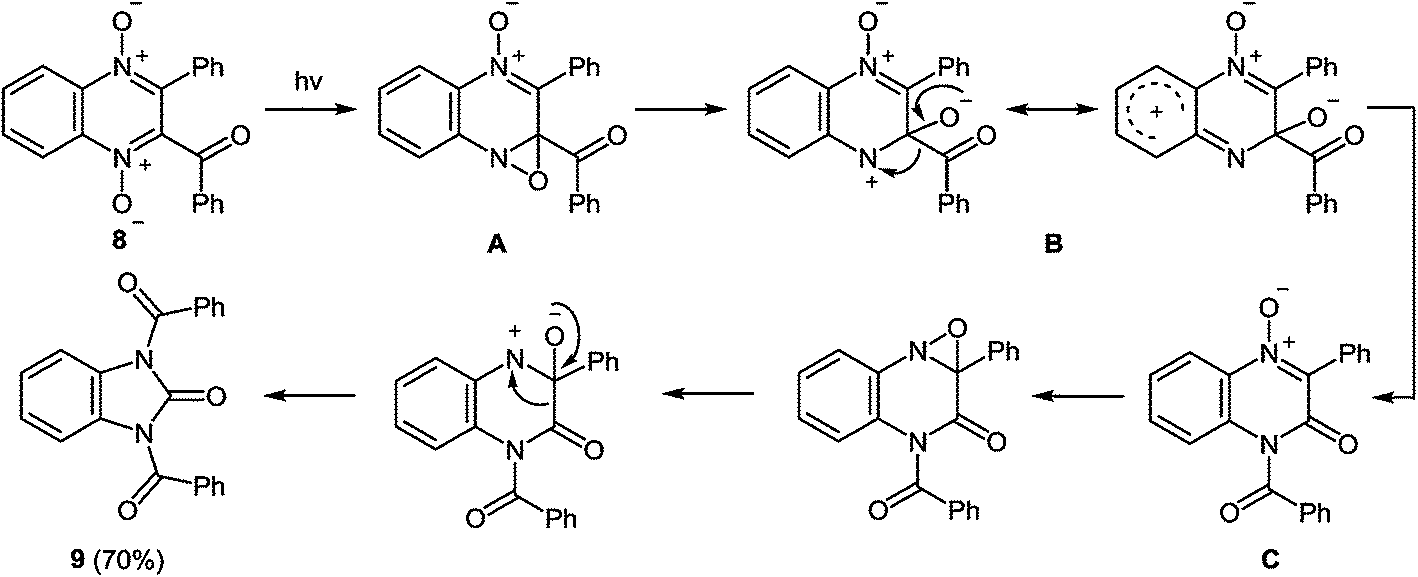 | ||
| Scheme 5 Proposed mechanism of the rearrangement of 2-benzoyl-3-phenylquinoxaline-di-N-oxide when exposed to sunlight. | ||
Ring contraction also occurs on irradiation of quinoxaline di-N-oxide of the type 10, the product of the reaction being a 1,3-disubstituted benzimidazolone 11 (Scheme 6).51b
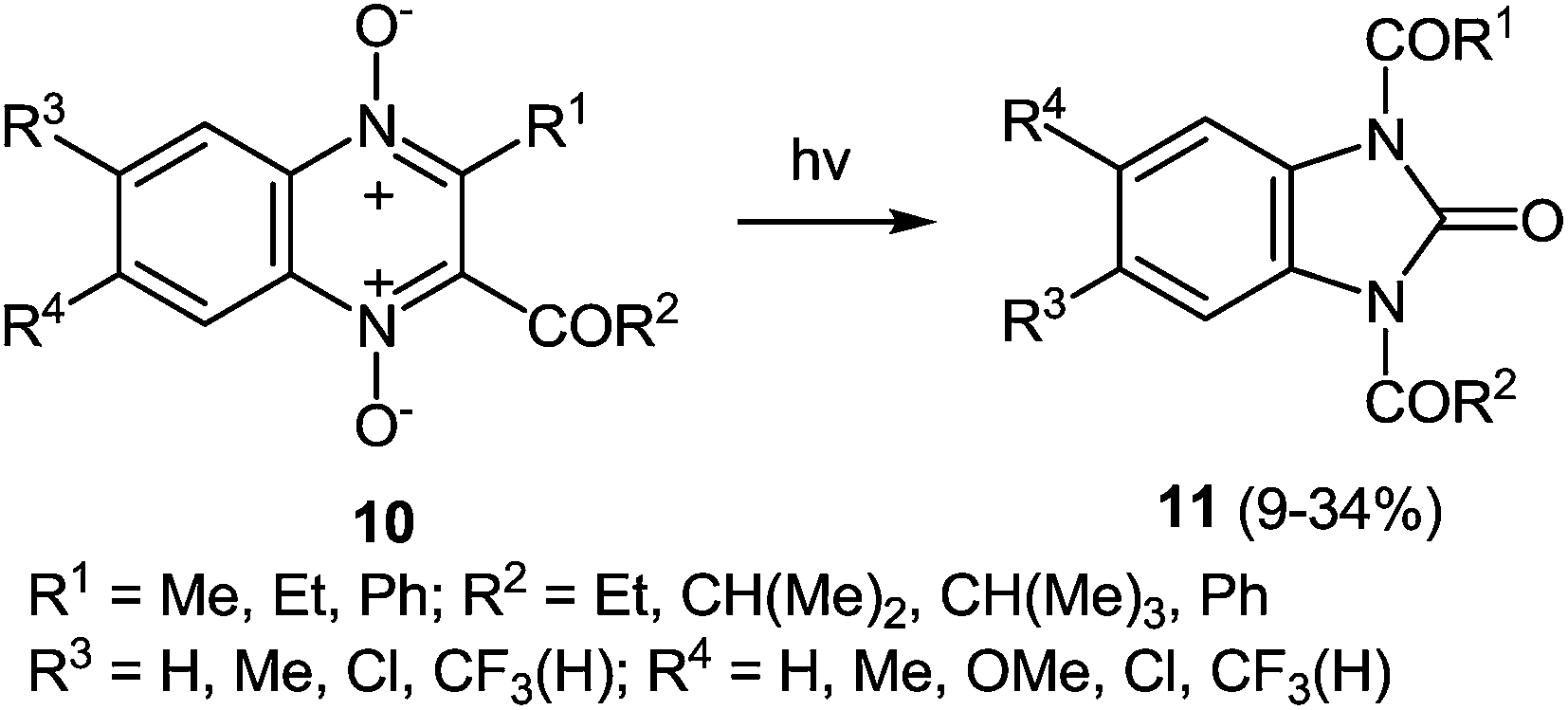 | ||
| Scheme 6 Ring contraction of quinoxaline-di-N-oxide when exposed to light. | ||
For example, when 3-hydroxy-2-phenyl- (12a), 7-ethoxy-3-hydroxy-2-phenyl- (12b) and 3-hydroxy-7-methyl-2-phenyl-quinoxaline-1-oxide (12d) are heated with acetic anhydride under reflux for 4 h, they yield l-acetyl-3-benzoyl-2-benzimidazolone (13a) and its 5-ethoxy- and 5-methyl-l-acetyl-3-benzoyl derivatives 13b and 13d, respectively. The latter on hydrolysis with aqueous alkali lose their acetyl and benzoyl groups and yield benzimidazolone 16a and its 5-ethoxy- (16b) and 5-methyl- (16d) derivatives (Scheme 7). 3-Hydroxy-2-(4-nitrophenyl)quinoxaline-1-oxide 12g remained unchanged even on prolonged heating under reflux with acetic anhydride. However, when the two reactants are heated together in a sealed tube at 180 °C for 12 h, the products of the reaction are 1,3-diacetyl- and l-acetyl-2(3H)-benzimidazolones 14a and 15 with 4-nitrobensoic acid. Similarly, 2-cyano-3-hydroxyquinoxaline 1-oxide 12i failed to react with acetic anhydride under ordinary conditions of reflux. However, in a sealed tube at 180 °C, 1,3-diacetyl-2-benzimidazolone 14a was produced as well.
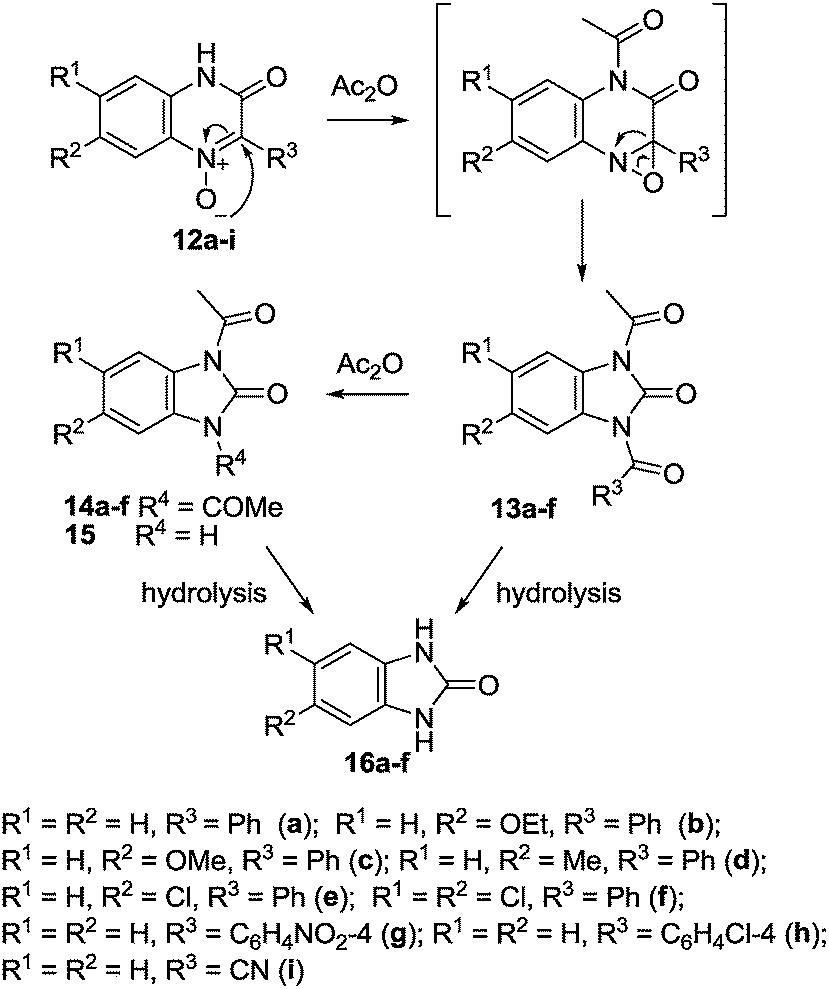 | ||
| Scheme 7 Proposed mechanism of the rearrangement of 3-hydroxy-2-R-quinoxaline-1-oxides when heated under reflux with acetic anhydride. | ||
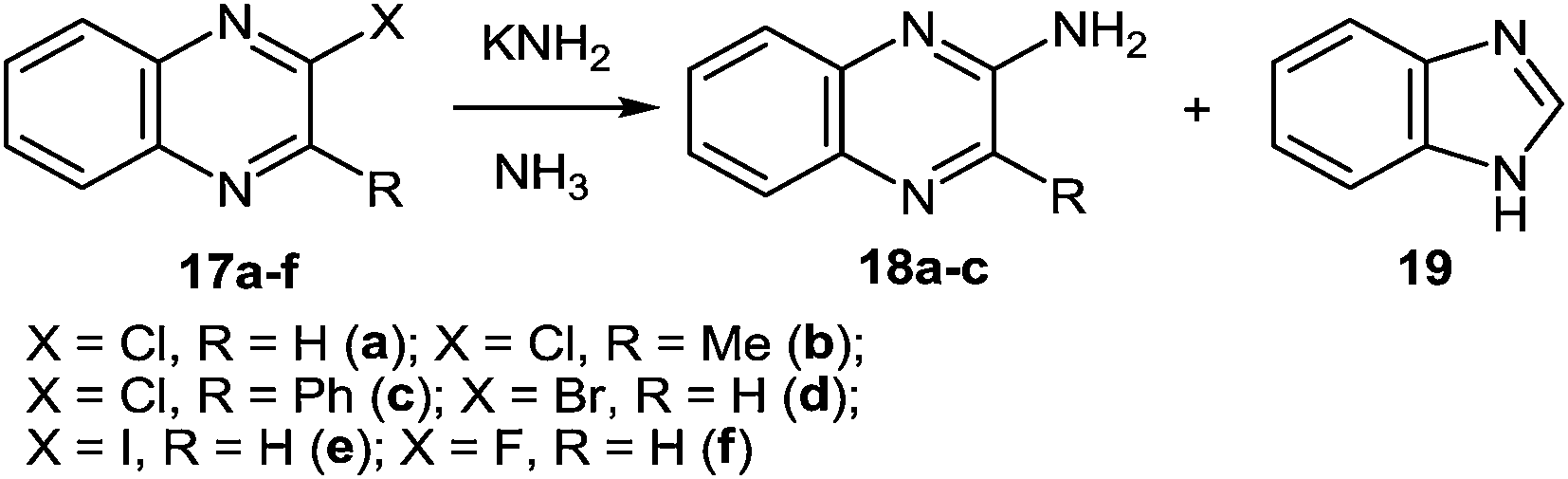 | ||
| Scheme 8 The reaction of 2-haloquinoxalines with potassium amide. | ||
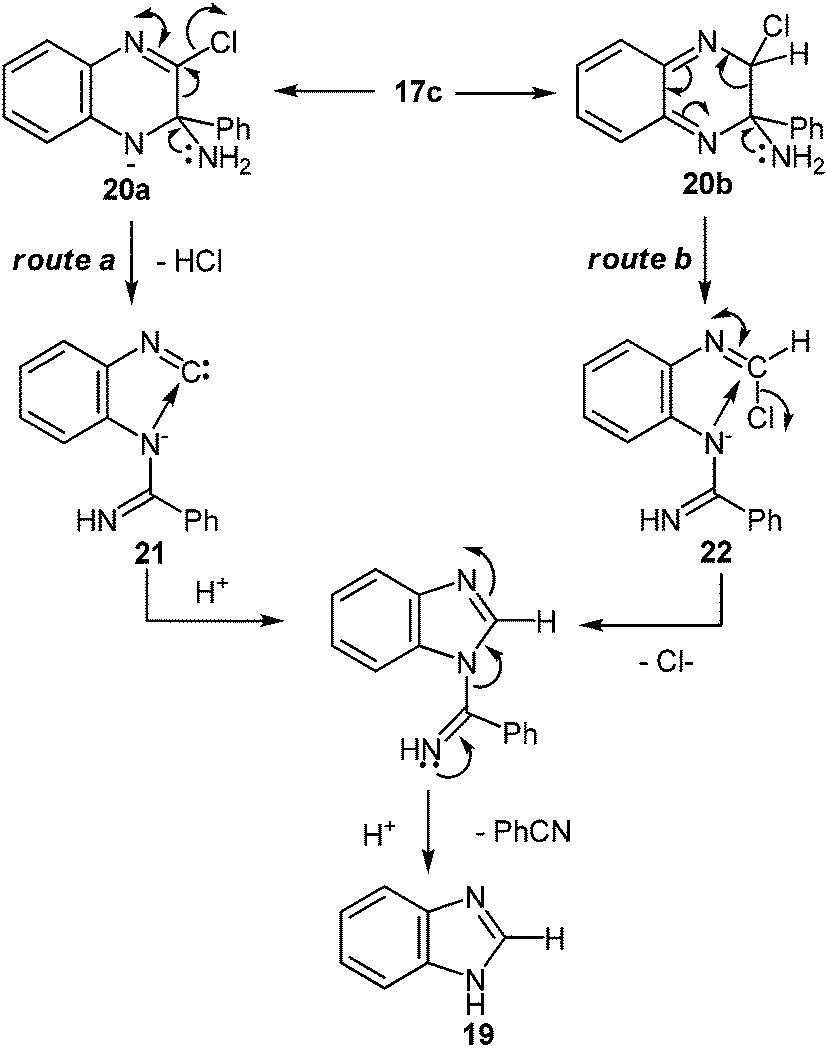 | ||
| Scheme 9 Proposed mechanism of the rearrangement of 2-chloroquinoxalines into benzimidazole when exposed to potassium amide in liquid ammonia. | ||
At present, it is yet unclear whether isonitrile 21 (Scheme 9, route a) or iminochloride 22 (Scheme 9, route b) is involved in the course of this reaction. It is tentatively suggested that this ring contraction occurs as shown below (Scheme 9). The formation of a nitrile has been confirmed by the fact that with 17a it is possible to isolate benzamidine, which is probably formed by the addition of potassium amide to benzonitrile.
It should be pointed out that with 2-chloroquinoxaline the main product is benzimidazole, and only some 2-aminoquinoxaline is formed. Under these conditions 2-bromo- (17d) and 2-iodo- (17e) quinoxaline are almost exclusively converted into benzimidazole, whereas with 2-fluoroquinoxaline 17f only a trace of benzimidazole is formed.54
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of 24, resulting in the 3,4-addition analogues to addition reactions in 6- and 7-oxopteridines with nucleophilic reagents.56 As above, the ring contraction into 25, and the reductive conversion of the –HCN–NH2 into a methyl group by a Wolff–Kishner type process, together with dehydration, produced 26 (Scheme 10).
N bond of 24, resulting in the 3,4-addition analogues to addition reactions in 6- and 7-oxopteridines with nucleophilic reagents.56 As above, the ring contraction into 25, and the reductive conversion of the –HCN–NH2 into a methyl group by a Wolff–Kishner type process, together with dehydration, produced 26 (Scheme 10).
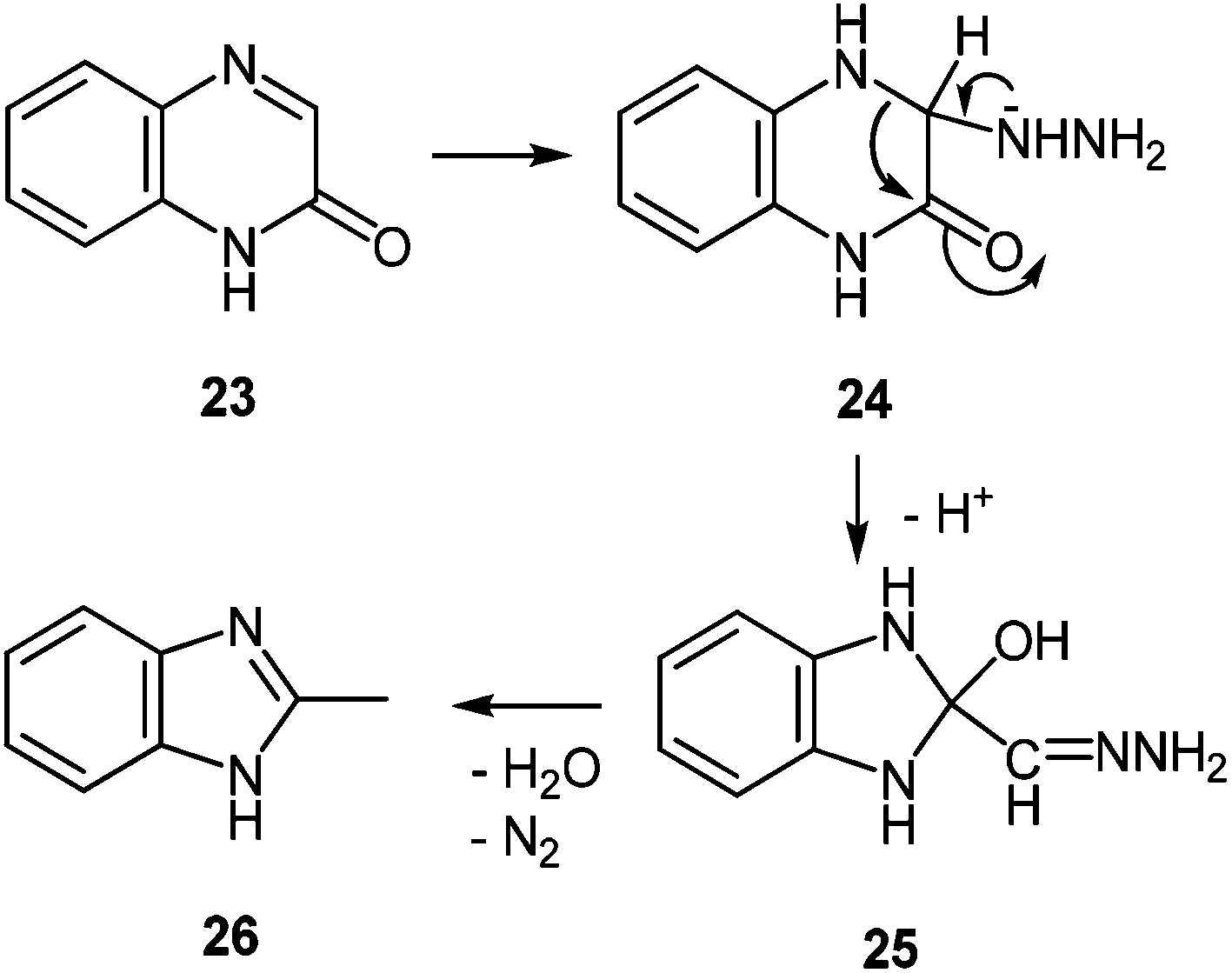 | ||
| Scheme 10 Hydrazinolysis of quinoxalin-2(1H)-one. | ||
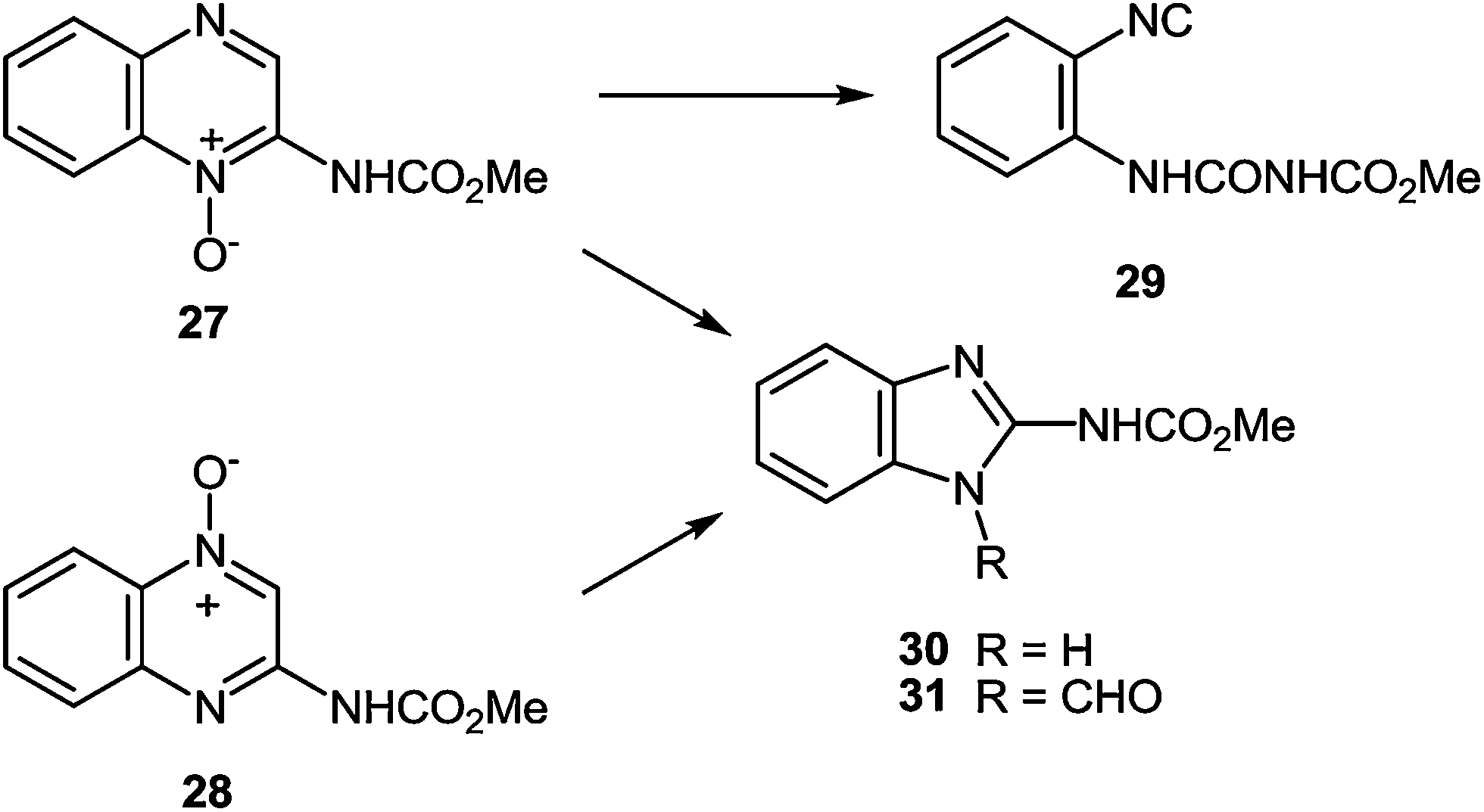 | ||
| Scheme 11 Photolysis of the quinoxalin-2-ylcarbamate-N-oxides. | ||
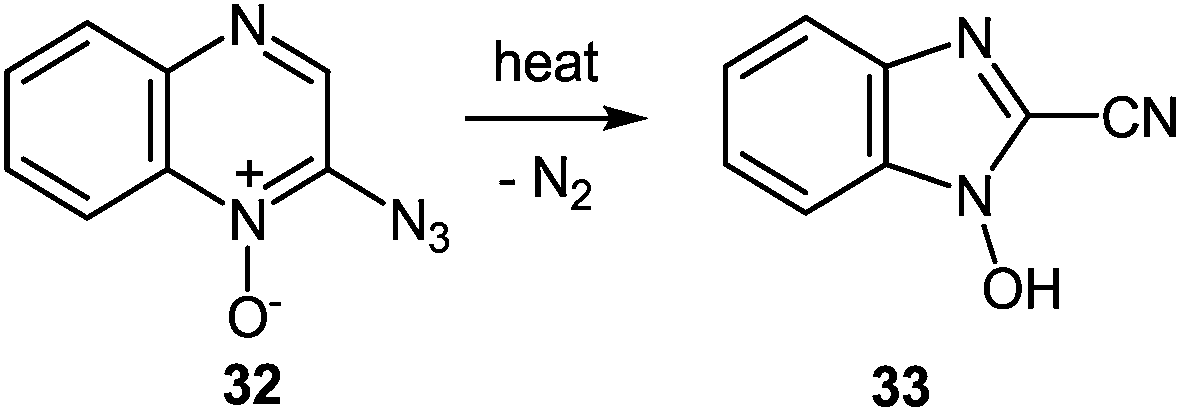 | ||
| Scheme 12 Thermolysis of 2-azidoquinoxaline-1-oxide. | ||
Oxidation of 2-phenylquinoxaline-4-oxide 34 with a 30% aqueous hydrogen peroxide in acetic acid59 or formic acid60 results in the 1,4-dioxide, which on reduction with sulfurous acid in methanol yields 2-phenylquinoxaline-1-oxide. However, treatment of the 4-oxide 34 with a 30% aqueous hydrogen peroxide in methanol and the presence of potassium hydroxide brings about 2-phenylbenzimidazole-3-oxide 35. This is a general reaction of 2-alkyl- and 2-alkoxyquinoxaline 4-oxide (Scheme 13).61
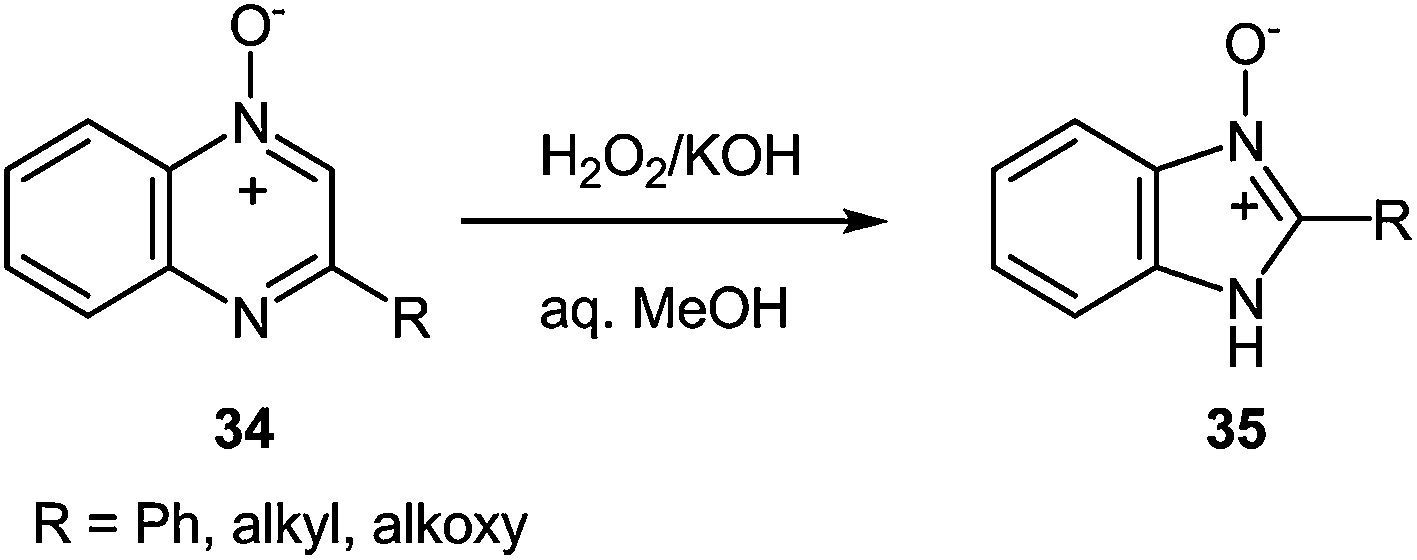 | ||
| Scheme 13 Oxidation of quinoxaline-4-oxides. | ||
As evident from the above eight examples of recycling of quinoxaline derivatives, the first two and the sixth one lead to the 2-substituted benzimidazoles, and the third and fourth ones result in the N-substituted benzimidazolones. All of them except the last one are special cases, i.e. implemented as the special representatives of the benzimidazole derivatives. As for the fourth example, the possibility of this reaction is limited to the synthesis of the derivatives of benzimidazolones with substituents only on the benzene ring. As a result of rearrangement they are eliminated as substituents at position 3 of the starting compounds. Thus, none of these methods of the synthesis of benzimidazole derivatives can compete with the classical Phillips–Ladenburg and Weidenhagen reactions.
2.2 Principles of the method
Quinoxalin-2(1H)-ones 37 can be converted into substituted benzimidazoles 40 following the reaction sequence, first reported in 2000,62 and shown in Scheme 14. Firstly, 3-α-chloro (or bromo)benzylquinoxalin-2(1H)-ones 36 are oxidized with the Kornblum type reactions63 or the direct oxidation of 3-benzyl (or alkyl) quinoxalin-2(1H)-ones 36 with the help of GrO3 in acetic acid with water62d,64 to give 3-aroyl- or 3-alkanoylquinoxalin-2(1H)-ones 37. 3-Aroyl- or 3-alkanoylquinoxalin-2(1H)-ones 37 react with 1,2-DABs 5 to give the spiro-quinoxalinone derivative 38. The spiro-quinoxalinone derivative 38 is then heated in acetic acid to give the benzimidazole derivative 40 through the proceeding cascade reactions involving: (a) acid-catalyzed ring-opening of spiro-compound 38 with the formation of quinoxaline derivative 39, (b) the intramolecular nucleophilic attack by the amino moiety on the carbonyl group leading to the formation of the final product 40 with the elimination of water. Practically for every case the spiro-compound 38 can be isolated and individually characterized, but the quinoxaline derivative 39 can only be rarely isolated. The sequence is thus based on the combination of the following facts: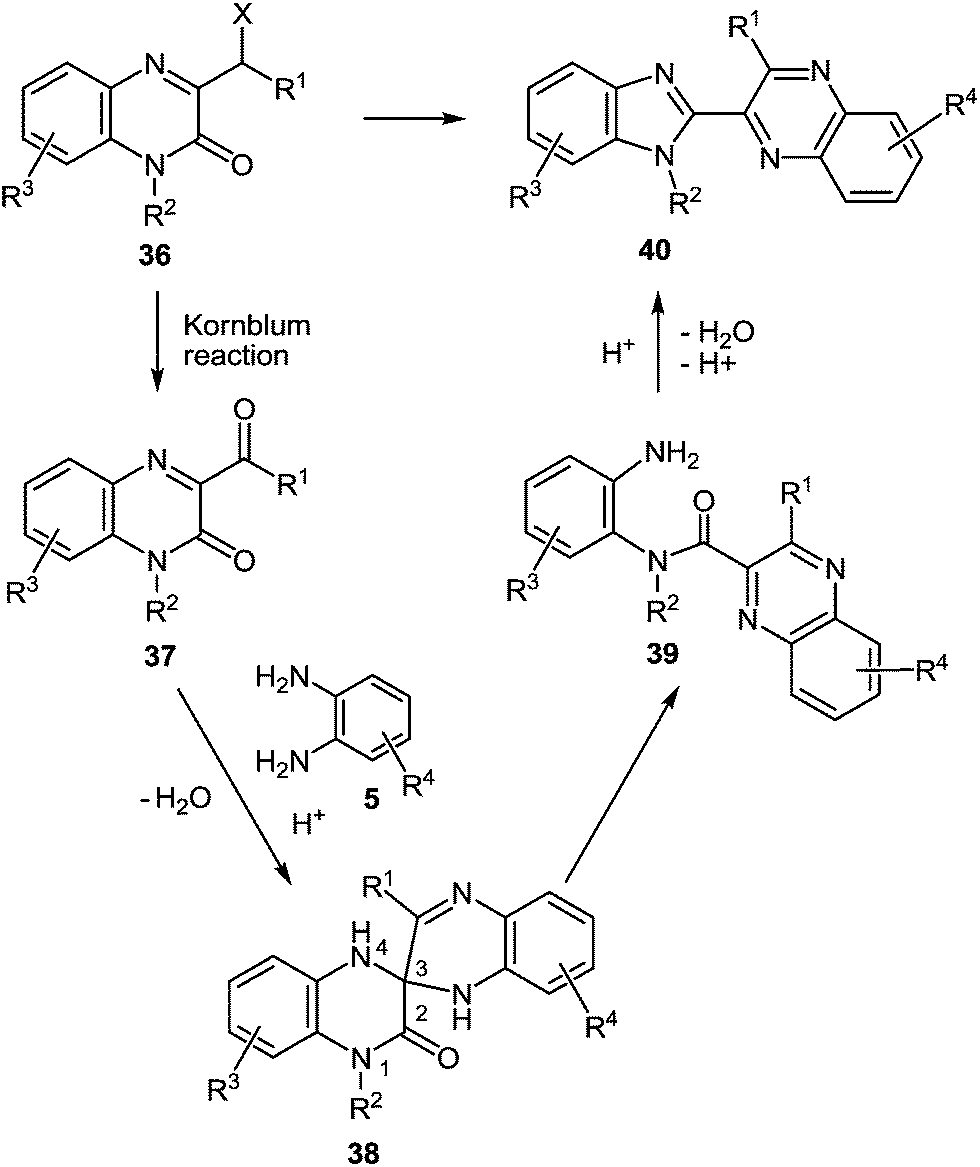 | ||
| Scheme 14 The reaction sequence of the rearrangement. | ||
(i) The presence of the carbonyl group at position 3 of quinoxalin-2(1H)-ones 37, according to the principle of Ogg and Bergstrom48 allows to consider them as the hetero analogues of α-diketones (see Sections 4 and 10).
(ii) The susceptibility of these systems to the reactions of the usual α-diketones, according to the Hinsberg reaction65 involves spiro-quinoxaline derivatives with at least one mobile hydrogen atom in the spiro-forming component.62
(iii) The susceptibility of the spiro-quinoxaline derivatives 38 towards the acid-catalyzed ring-opening with the formation of quinoxaline derivatives 39, and the intramolecular ring closure reaction with the formation of benzimidazole derivatives 40.
In the reactions above aroyl- and alkanoylquinoxalinones were dealt with as hetero analogues of α-diketones. Accordingly, we assume the 3-(α-haloalkyl)- and 3-(α-halobenzyl)quinoxalin-2(1H)-ones, 3-(α-aminobenzyl)quinoxalin-2(1H)-ones and 3-arylacylidene-3,4-dihydroquinoxalin-2(1H)-ones to be analogues of α-haloketones, α-aminoketones and β-diketones, respectively.
2.3 Advantages of the method
The present method for the synthesis of substituted benzimidazoles starting from quinoxalin-2(1H)-one derivatives has the following characteristics or distinct advantages over the previously used routes (the Phillips–Ladenburg and the Weidenhagen reactions) or their numerous variations.(A) Firstly, the acid-catalyzed rearrangement of spiro-quinoxalinone derivative 38 through the o-aminoanylide quinoxaline 2-carboxylic acid 39 involves a milder reaction condition and provides almost quantitative yields of the benzimidazole derivative 40 (Scheme 14). Most of the popular approaches generally involve the condensation of an arylenediamine with a carbonyl equivalent (Scheme 15). For example, the reaction of 1,2-DABs with carboxylic acid or acid chloride results in intermediate amide 43. In order to produce benzimidazole 46 the latter in turn could undergo a cyclodehydration reaction at elevated temperatures under strong acidic or alternatively under harsh dehydrating conditions, (Scheme 15, route a). Similarly, esters, lactones and anhydrides could generate benzimidazoles via the cyclization of amide 43, although given the rather harsh reaction conditions required and the poor diversity profile of the final products their scope might be limited. For instance, the reaction of 1,2-DABs with aliphatic esters and lactones involves the use of strong mineral acids such as hydrochloric acid, sulfuric acid, hot glacial acetic acid or polyphosphoric acid under very high temperatures, i.e. the conditions are not absolutely compatible with a broad range of functional groups and desirable substrates. Aromatic esters require temperatures of up to 250–300 °C, thus rendering the synthesis of 2-arylbenzimidazoles almost impractical.46a However, the reaction of aromatic esters with 1,2-DABs under the Weinreb conditions could provide access to 2-arylbenzimidazoles.46b In the class of acid anhydrides of monobasic acids, only acetic anhydride has been practically used in the preparation of 2-methylbenzimidazoles. Cyclic anhydrides of dibasic acids have also been used in the synthesis of benzimidazoles, although high temperatures and strong acids are usually necessary to convert the intermediate N-(o-aminophenyl)-imide into the desired benzimidazole.47a,b Besides, a mixture of regioisomeric benzimidazoles could result from the reaction of nonsymmetric anhydrides with arylenediamines.
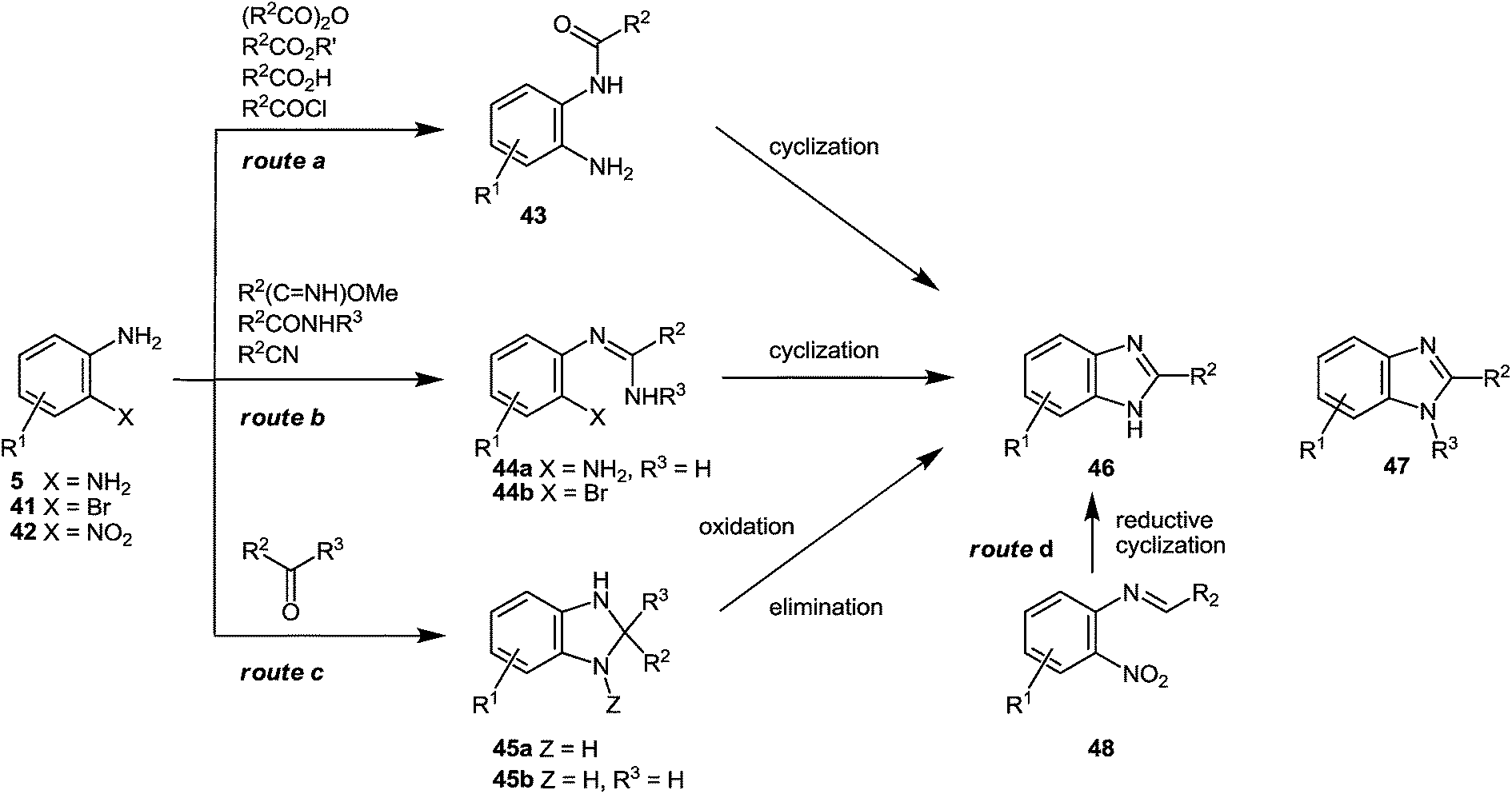 | ||
| Scheme 15 Common strategies for the synthesis of benzimidazoles. | ||
In the presence of HCl the reaction of 1,2-DABs with amides66 and nitriles67 at 200–250 °C could also afford 2-substituted benzimidazoles with the general structure 46via the cyclization of intermediate amidine 44a (Scheme 15, route b). Alternatively, upon the formation and subsequent cyclization of amidine 44a under milder conditions68 the reaction of 1,2-DABs with an imidate could afford benzimidazole 46 as well. Although the imidate route could provide access to a diverse set of 2-substituted benzimidazoles starting from several commercially available aliphatic and aromatic nitriles, the hydroscopic nature of the intermediate imidates might be of concern, particularly in a high throughput set-up. The palladium catalyzed intramolecular N-arylation reaction of the o-bromophenylamidine precursors of type 44b, resulting from the assisted POCl3 condensation of bromoaniline 41 and an amide, has been recently developed providing entry to N-substituted benzimidazoles with the general structure 47.69 Despite the somewhat harsh conditions required to generate the intermediate amidine precursors, this method proves successful in the regioselective synthesis of N-substituted benzimidazoles which is a current limitation as regards many other approaches.
Aldehydes and, to a lesser extent, ketones can also afford benzimidazoles when condensed with 1,2-DABs (Scheme 15, route c). Although the reaction of ketones with 1,2-DABs in the presence of HCl at 250–300 °C can yield benzimidazole 46 due to the aromatization of intermediate benzimidazoline 45a, their use has been rather limited.1b,70 Furthermore, since the aromatization of benzimidazoline 45a occurs via the elimination of an alkyl group, a mixture of benzimidazoles could result from non-symmetric ketones. Alternatively, aldehydes have been used more extensively in the preparation of 2-substituted benzimidazoles according to the Weidenhagen's method.71
For example, condensation of 1,2-DABs with an aldehyde, followed by the oxidation of the intermediate benzimidazoline 45b could afford benzimidazole 46 as well. While the oxidation can proceed spontaneously by disproportionation, this can lead to some of side products. Oxidative methods usually require heating in nitrobenzene or DMF at elevated temperatures, as well as the use of metal ions, iodine, organic oxidants or inorganic sulfites when heated.72 However, for the synthesis of N-substituted and N–H benzimidazoles one mild set of oxidation conditions utilizing oxone has been recently described.72
Method of reductive cyclization of N-benzylidene-2-nitroanilines 48 (R2 = Ar), prepared from o-nitroaniline 42 and benzaldehydes (Scheme 15, route d) could be a viable alternative to the widely used 1,2-DAB based synthetic methods. But according to our knowledge, it was not as yet been studied to date. Triethylphosphite,73 triruthenium dodecacarbonyl74 in the presence of carbon monoxide, and recently, phenylmagnesium chloride75a have been successfully utilized as the reducing agents for this transformation. The reaction presumably proceeds via an in situ aryl nitro reduction, followed by an intramolecular cyclization,75 and results in benzimidazole 46 (R2 = Ar). This strategy requires the preparation and isolation of the corresponding N-benzylidene-2-nitroanilines 48 before subjecting them to cyclization conditions though it could obviate the preparation and isolation of the intermediate 1,2-DABs in particular those that are water-soluble or prone to air-oxidation.
(B) Secondly, the synthesis of substituted benzimidazoles can be accomplished starting from the quinoxalin-2(1H)-one derivative 36 easily available with the use of various preparatively simple ways under mild conditions from corresponding substituted 1,2-DABs and pyruvates. In doing so as a result of the rearrangement substituents of the benzene ring of the quinoxalinone system appeared to be on the benzene ring of the benzimidazole ring. As a result of the rearrangement the substituent R2 at position N(1) of the quinoxalin-2(1H)-one system are transferred to the N atom of benzimidazole system.
(C) The route 36 to 40 clearly shows that a benzyl (or haloalkyl) group can be converted (by use of the Kornblum reaction) into a ketone group necessary for the introducing of quinoxaline ring in to position 2 of quinoxalinone 37 according to the Hinsberg reaction.65 In this reaction compound 37 formally acts as the hetero analogue of α-diketone.
(D) As can be seen from the reaction depicted in Scheme 14, the key step in this case is the formation of spiro-compound 38. As a result of the cascade reaction this compound is transformed into a benzimidazole derivative 40, with a heterocyclic system. This system acts as a spiro-fragment in the intermediate compound 38. The result makes it possible to propose the main principle for this rearrangement. Any of the spiro-derivatives of 1,2,3,4-tetrahydroquinoxalin-3-one with at least one mobile hydrogen atom in their spiro-forming components can be converted into the benzimidazole derivative with the spiro-forming component at position 2 (Scheme 16).76
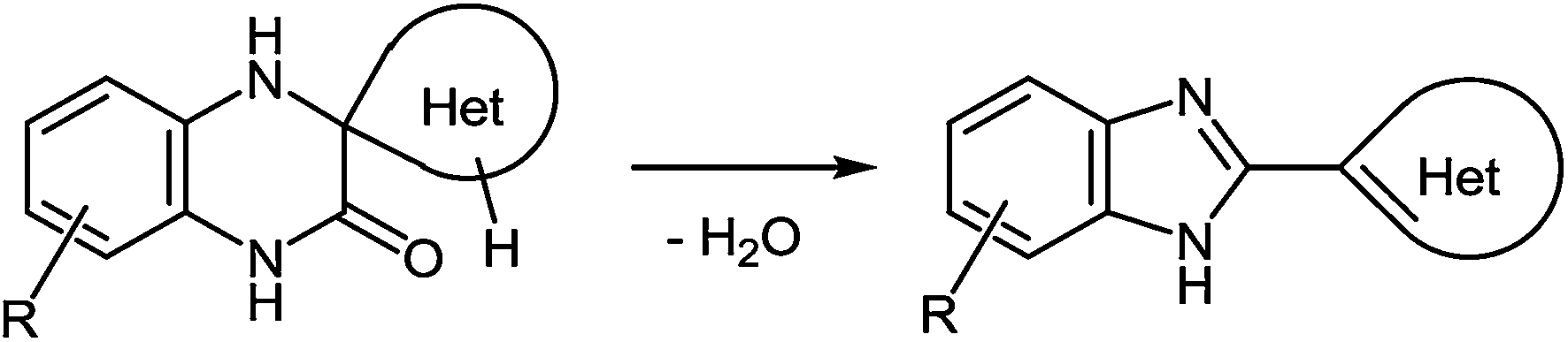 | ||
| Scheme 16 Schematical presentation of the rearrangement. | ||
(E) The understanding of the reaction mechanism makes it possible for us to make a bold assumption as seen from Fig. 4, all the reactions of aroyl- and alkanoylquinoxalines when interacting with 1,2-DABs at the initial stage behave as hetero analogues of α-diketones, i.e., as iminoketones (see Sections 4 and 10). Then the problem appears, why other quinoxaline derivatives with certain substituents do not behave like α-haloketones 49 (see Section 5), β-diketones 50 (see Section 6), α-aminoketones 51 (see Section 7), methyl ketones 52 (see Section 8), o-aminoaldehydes (or ketones) 53 (see Section 9), etc.
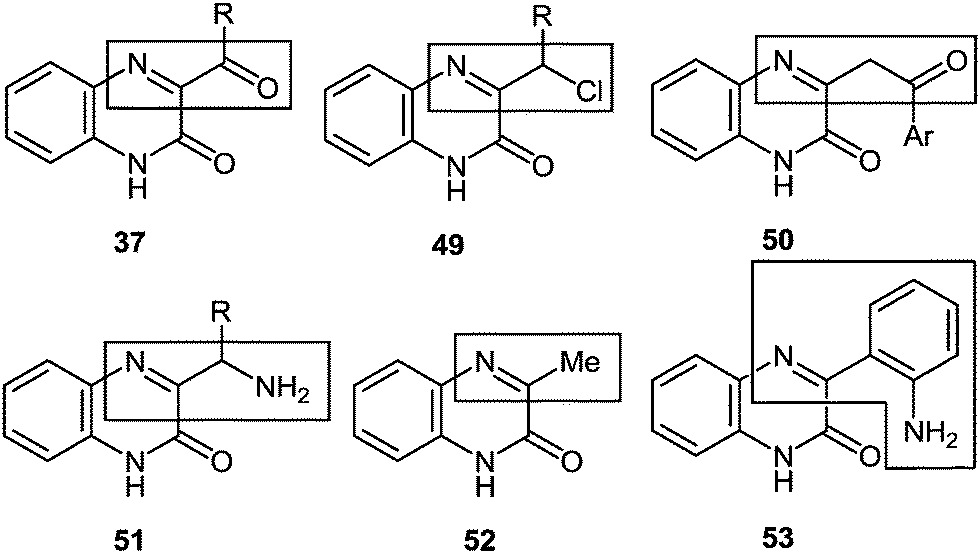 | ||
| Fig. 4 Schematical presentation of the quinoxalin-2-ones as functional ketones. | ||
3 Synthesis of 2-(benzimidazol-2-yl)quinoxalines
3.1 From glycerol and 1,2-diaminobenzenes with use of gold catalysis
Methods for the synthesis of these hetarylquinoxalines are limited. An option involves the reaction of the 1,2-DAB 5a with 3-hydroxyimino-2-butanone 54 and bromine in a one-pot reaction (Scheme 17a).77 Another possibility is to react 1,2-DABs 5a–g with quinoxalin-2-carboxylic acid 56, but this synthetic route needs to be performed in a polyphosphoric acid media at 200 °C (Scheme 17b).78 A third route is to react the 1,2-DAB 5a with quinoxalin-2-carboxaldehyde 58 in benzene at reflux temperature (Scheme 17c).79 In all the three cases the yield of the target product remains very low.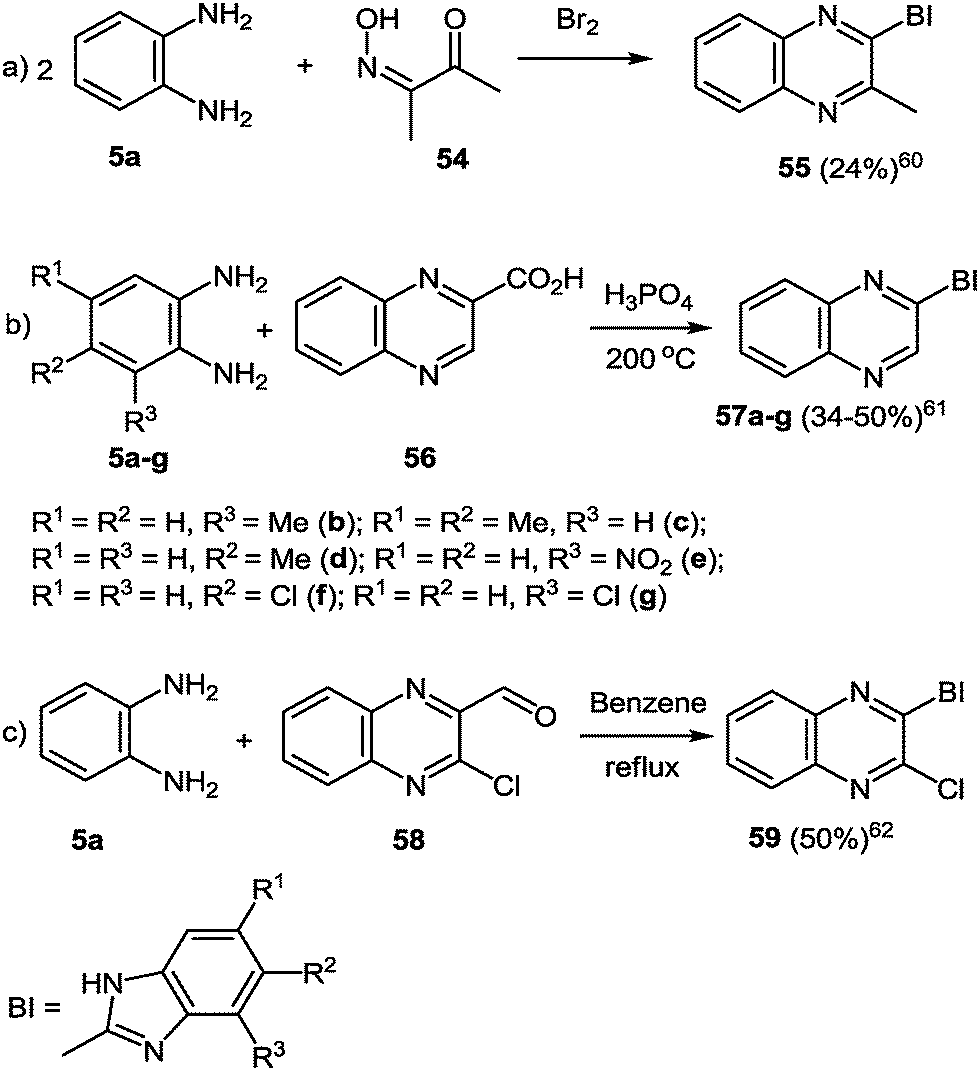 | ||
| Scheme 17 Benzimidazolylquinoxaline synthesis routes according to the ref. 60–62. | ||
A gold catalyst (Au/CeO2) was used to synthesize benzimidazolylquinoxalines by two novel methods in a multistep one-pot methodology.80 The first method involved the oxidative coupling of glycerol 60 with 1,2-DAB 5a performed at 140 °C using diglyme as a solvent and lead to the benzimidazolylquinoxaline compound 63a through the formation of intermediates 61a and 62a (Scheme 18). Herein, the benzimidazolylquinoxaline possesses the same substituents in both heterocycles. After that to expand the synthetic scope, an alternative route that allows combining different substituents in both heteroaromatic moieties was designed.
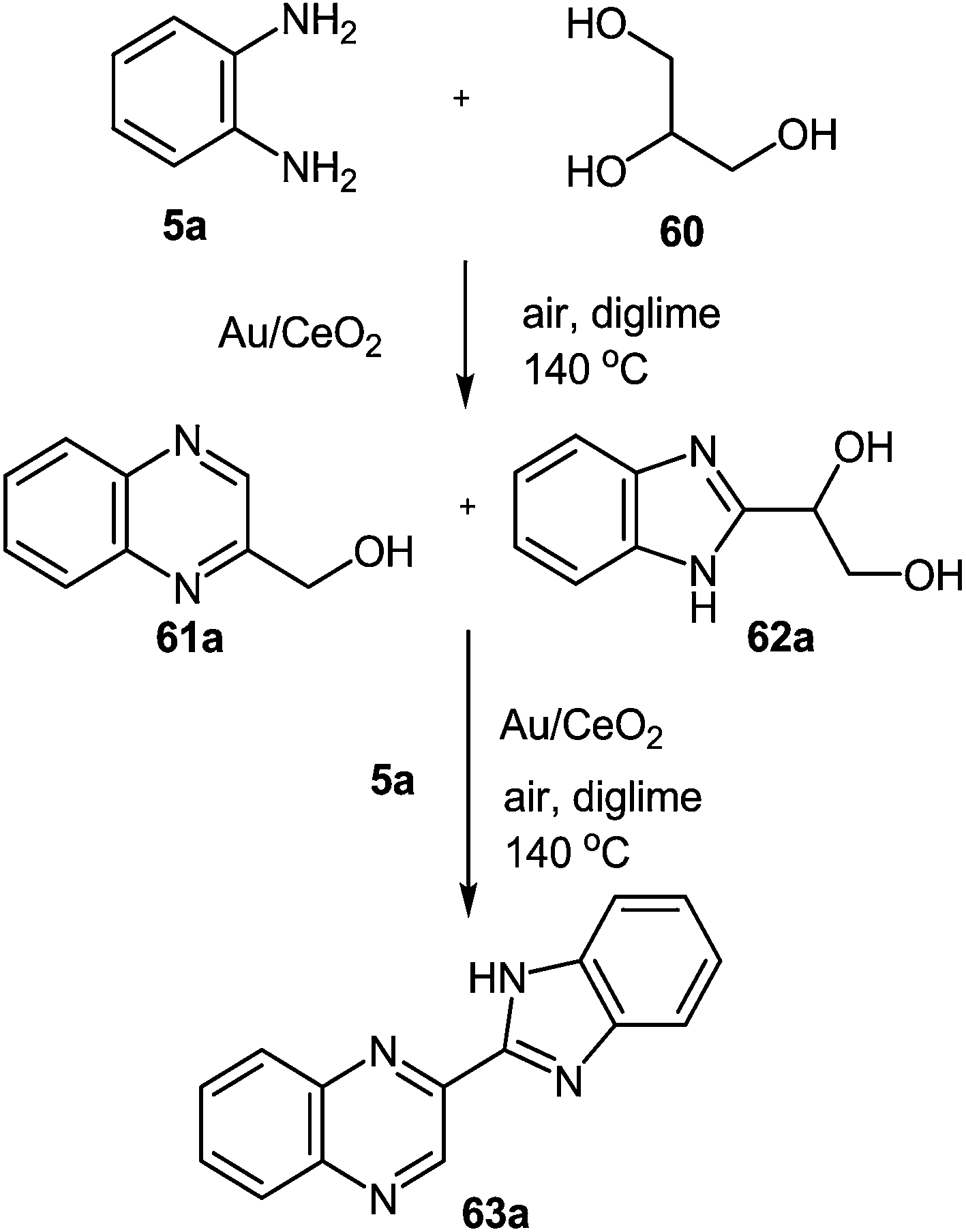 | ||
| Scheme 18 New one-pot two step process for the synthesis of benzimidazolylquinoxalines starting from glycerol. | ||
However, at the beginning of the reaction, the quinoxalin-2-ylmethanol 61a, formed by oxidative coupling between glycerol 60 and 1,2-DAB 5a, and the 1-(1H-benzo-[d]imidazol-2-yl)ethane-1,2-diol 62a, which could be produced through the oxidation of one of the primary alcohol groups of glycerol and subsequent coupling with 1,2-DAB 5a, were the predominant products (Scheme 18). Both compounds (61a and 62a) exhibited a primary and unstable character and after one hour of reaction, the concentrations of both intermediates began to decrease, due to their conversion into 2-(1H-benzo[d]imidazol-2-yl)quinoxaline 63a, produced through oxidation-cyclization of 61a and 62a with another 1,2-DAB molecule 5a. Other byproducts such as 1H-benzo[d]imidazole 64a, quinoxaline 65a, (1H-benzo[d]imidazol-2-yl)methanol 65a and 1H,1′H-2,2′-bibenzo[d]-imidazole 66a were also detected in the reaction media (Scheme 19). The proposed mechanism (Scheme 19) of the processes involves the oxidation of glycerol 60 to glyceraldehyde 68 and subsequently to the dicarbonyl compound 69.
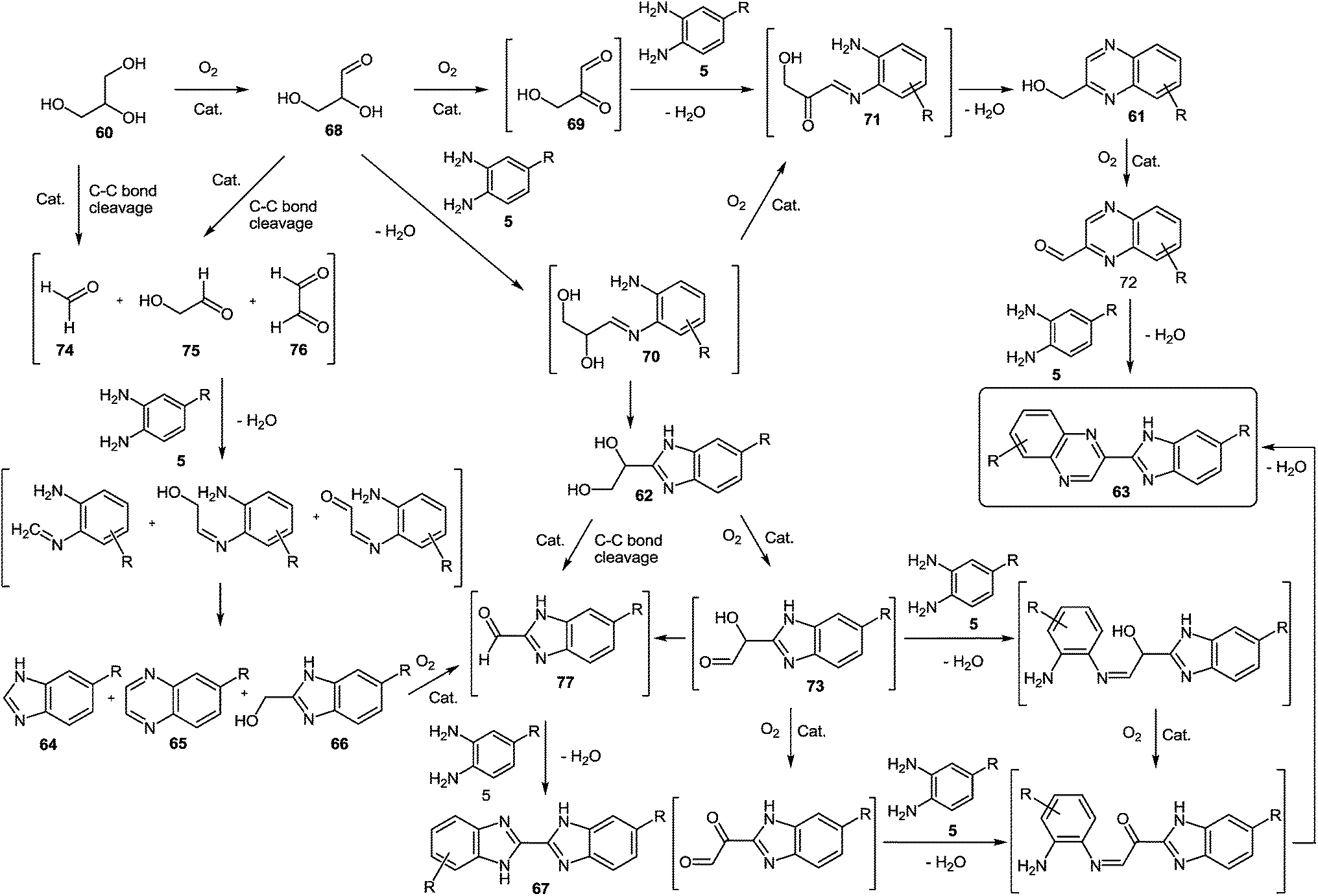 | ||
| Scheme 19 Proposed benzimidazolylquinoxalines synthesis mechanism starting from 1,2-DAB derivatives and glycerol. | ||
Both compounds can condense with 1,2-DAB to produce the imine intermediates 70 and 71, with 70 converted into product 71 by fast oxidation of the remaining hydroxyl group. Later, product 71 follows a condensation reaction to yield hydroxymethylquinoxaline intermediate 61, which can be further oxidized to 2-carboxaldehydequinoxaline 72, which couples with another molecule of 1,2-DAB to reach the benzimidazolylquinoxaline derivative 63. Besides, the imine intermediate 71 can result in the dihydroxybenzimidazole intermediate 62, which through a subsequent oxidative coupling with 1,2-DAB produces the benzimidazolylquinoxaline derivative 63. On the other hand, the formation of the byproducts detected by gas chromatography could be due to the oxidative cleavage of glycerol 60, glyceraldehyde 68, the dihydroxybenzimidazole intermediate 62, and the α-hydroxycarbonylbenzimidazole intermediate 73 into different carbonyl compounds such as 73, 74, 75, and 76. Later the coupling with 1,2-DAB produces the byproducts 64, 65, 66, and 67 through cyclization in minor amounts.
In Scheme 20, good yields of benzimidazolylquinoxaline derivatives were obtained. However, with electron-withdrawing substituents such as nitro, chloro or nitrile groups, the yields of quinoxaline derivatives were slightly lower with respect to those of 1,2-DAB or with respect to 1,2-DABs with electron-donating substituents such as methyl and methoxy. In all these cases the reactions proceed with the formation of regioisomers of benzimidazolylquinoxalines.
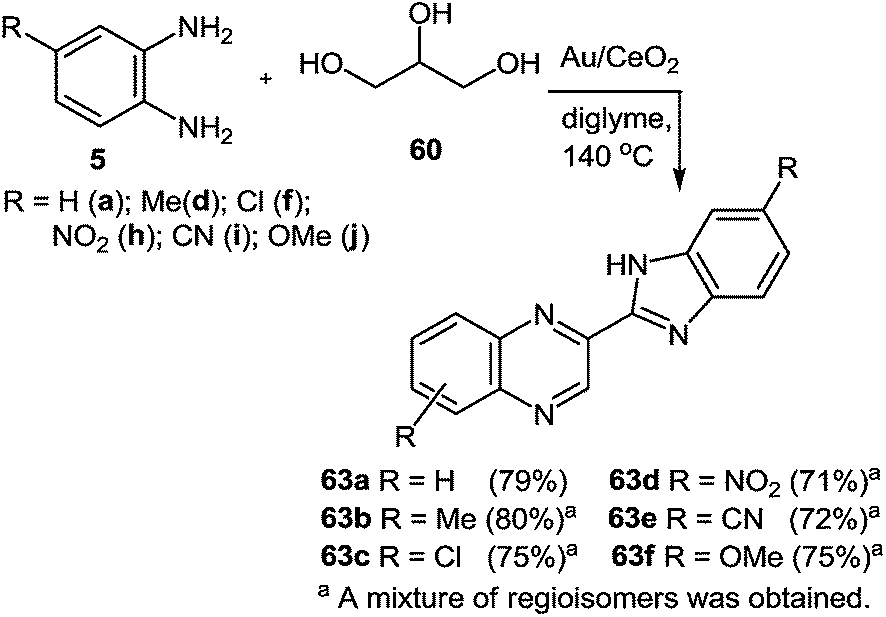 | ||
| Scheme 20 Synthesis of benzimidazolylquinoxaline derivatives starting from 1,2-DABs and glycerol with Au/CeO2 as a catalyst. | ||
To develop a method for the synthesis of the 2-benzimidazolylquinoxaline derivatives with different substituents in the benzimidazole and quinoxaline fragments the conditions for the formation of the assumed intermediate products of the reaction, i.e. hydroxymethylquinoxaline 61a, 1-(1H-benzo[d]imidazol-2-yl)ethane-1,2-diol 62a, and its protected derivative 80 were investigated (Scheme 21).80
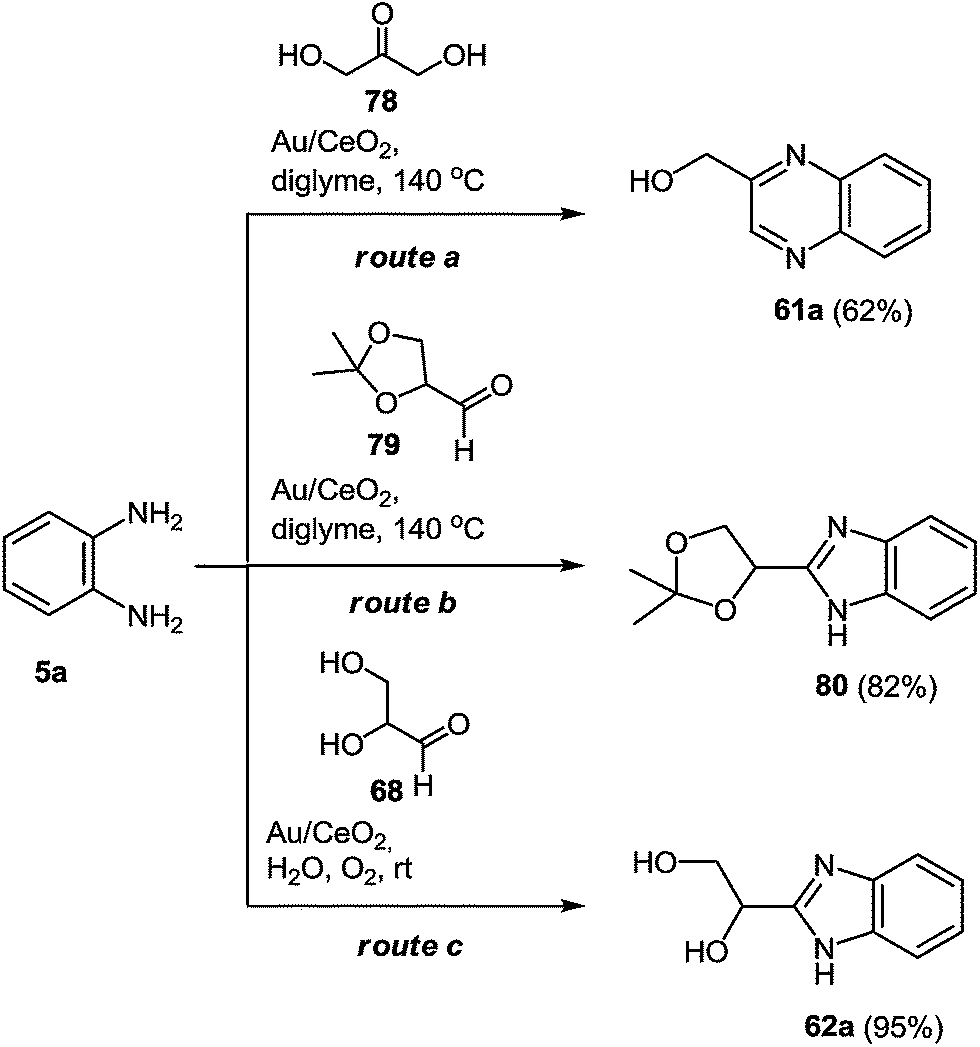 | ||
| Scheme 21 Possible synthetic routes to yield the intermediate quinoxaline 60a or benzimidazoles 79, 61a. | ||
Following the optimization of the reaction conditions with different substrates, the results presented in Scheme 22 showed that only the reaction between glyceraldehyde and 1,2-DAB (Scheme 21, route c) in the presence of Au/CeO2, oxygen pressure, and at room temperature, produced the benzimidazole intermediate 62 with a high selectivity with complete conversion. Consequently, the synthesis of the benzimidazole intermediate from glyceraldehyde and 1,2-DAB derivative was chosen as the optimum first step of the reaction.
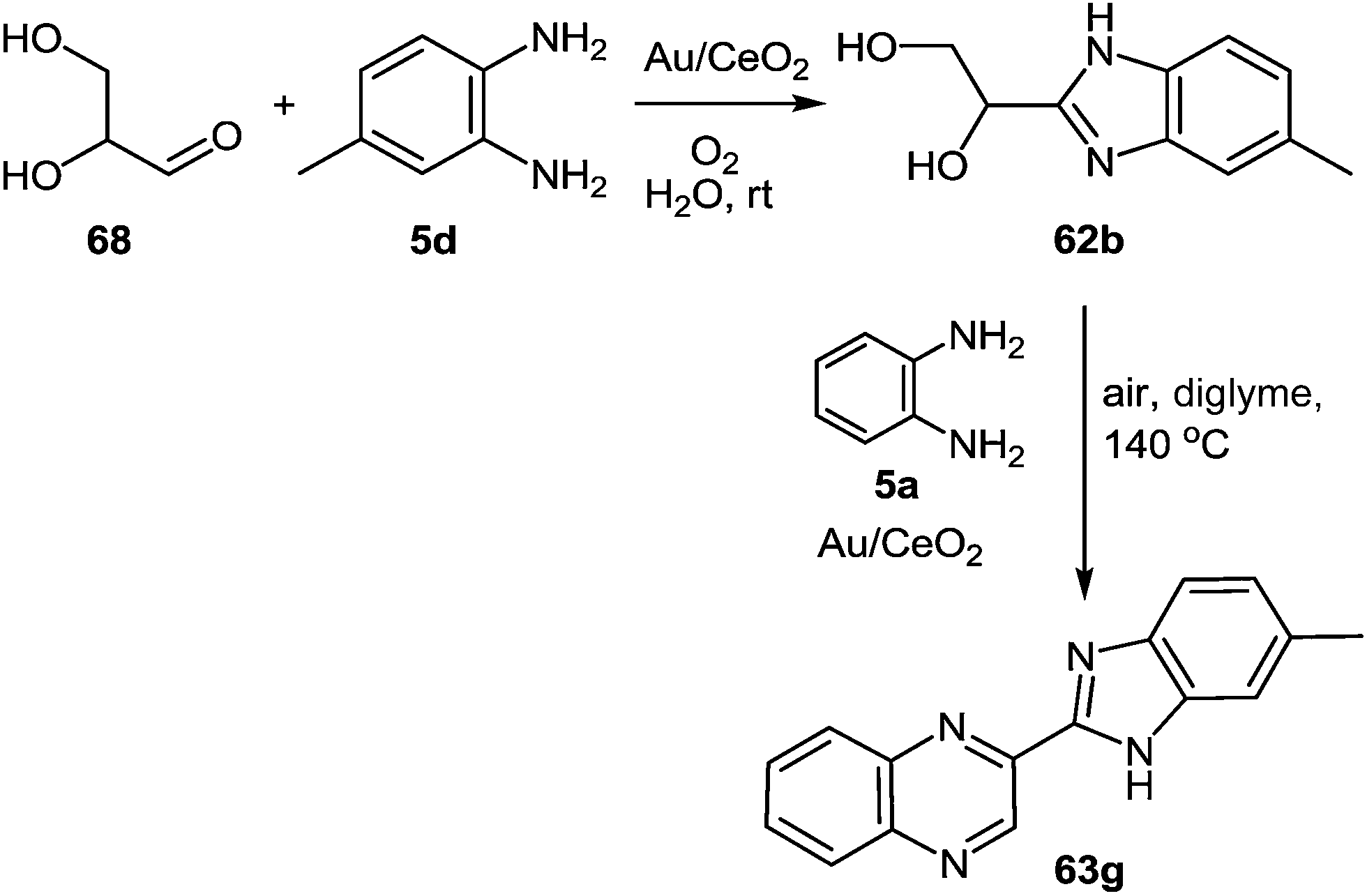 | ||
| Scheme 22 New one-pot two-step process to produce benzimidazolylquinoxalines starting from glyceraldehyde. | ||
This second process involves two sequential steps with very different reaction conditions. In the first one, glyceraldehyde is coupled with an 1,2-DAB derivative in the presence of Au/CeO2 to produce the intermediate 62 under very mild reaction conditions (room temperature, 3 bar O2, and water as a solvent). After the complete conversion of glyceraldehyde, a solution of the second 1,2-DAB molecule 5d in diglyme is added, and the temperature is increased to 140 °C while the water is removed by a Dean–Stark system. The main product observed under these conditions is the benzimidazolylquinoxalines 63g (Scheme 22), which is formed by oxidative coupling between the intermediate 62 and the 1,2-DAB molecule 5d.80 Both routes were applied to the synthesis of different benzimidazolylquinoxalines derivatives, obtained in yields between 60 and 80%.
As can be seen from the reaction mechanism (Scheme 23) and from the data in the Scheme 20, this method cannot be used effectively in the case of the synthesis of substituted derivatives of 2-benzimidazolylquinoxalines, as the reactions of mono-substituted derivatives of 1,2-DAB proceed with the formation of the mixtures of regioisomers, which are difficult to separate. The problem is further complicated with the diversely substituted derivatives of 1,2-DAB. Expensive (Au) and environmentally unsafe (CeO2) catalyst also limits the possibilities of this method.
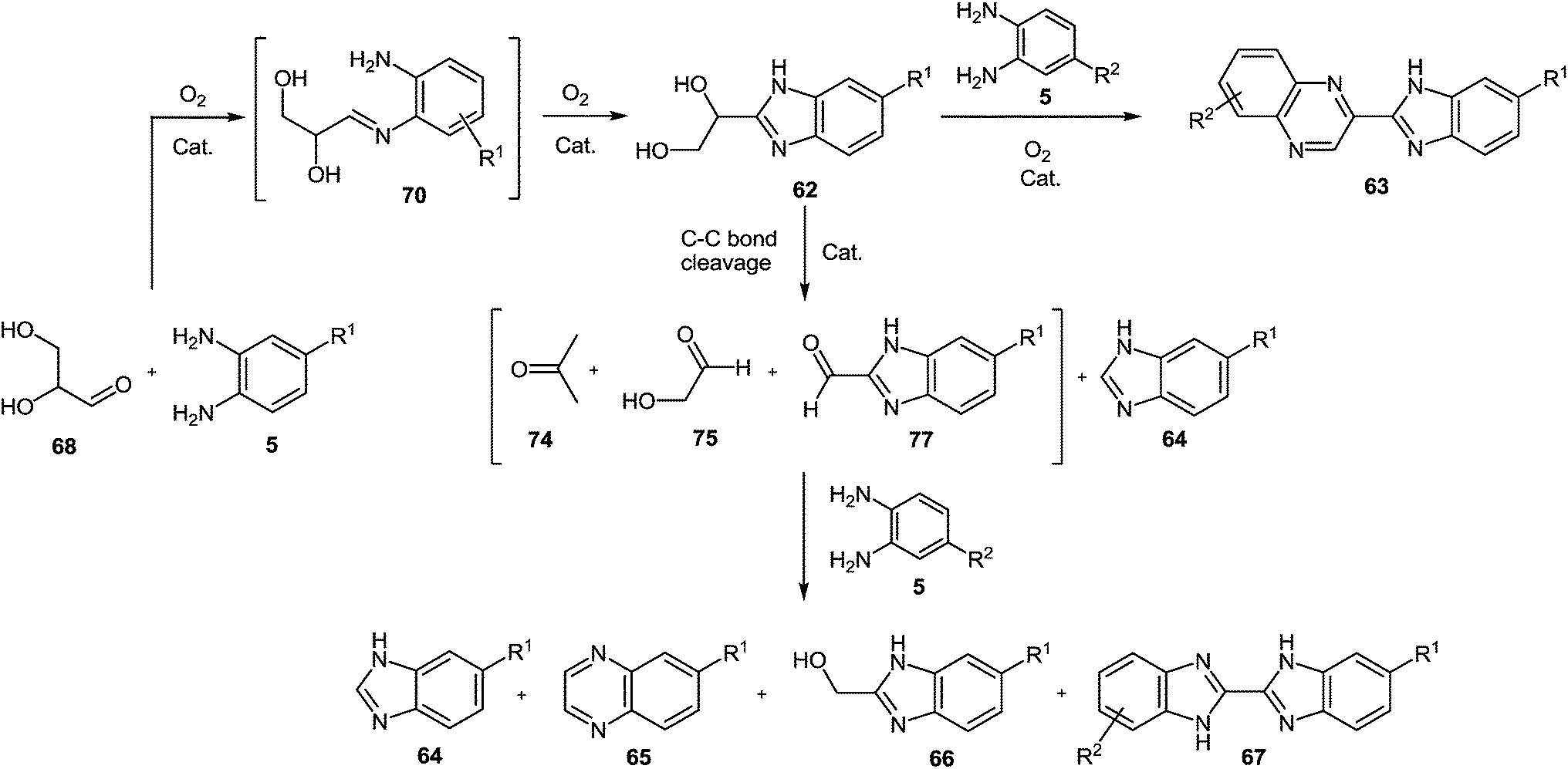 | ||
| Scheme 23 Proposed benzimidazolylquinoxalines synthesis mechanism from 1,2-DABs and glyceraldehyde. | ||
3.2 Via the isocyanide based multi-component reactions (IMCRs)
IMCRs are considered as a principal field of study for the generation of both new chemo type diversity and preferred methodologies to produce known heterocycles.81 In particular, the Ugi reaction, is extremely versatile enabling access to the numerous of small molecules through a various strategies that include post-condensation modifications of the Ugi adduct and use of variety of nucleophiles, which can trap nitrilium intermediate both intra- and intermolecularly (Scheme 24). Several groups have developed concise methodologies that enable access to diazepines,82 ketopiperazines,83 imidazolines,84 β-lactams85 hydantoins, etc.86 The final ring closure is accomplished through the amide bond formation in the Ugi/deprotect/cyclize strategies.81e,f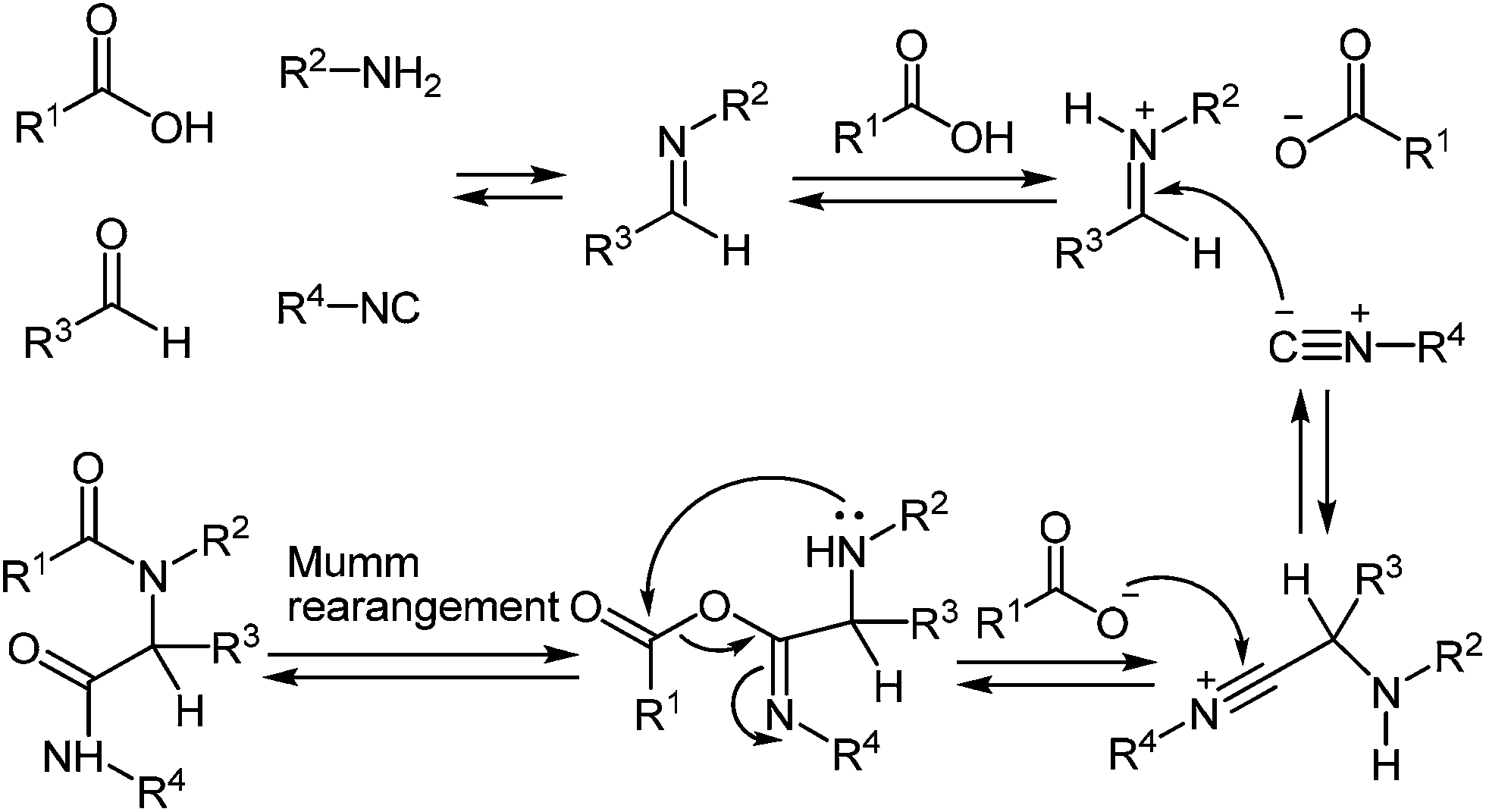 | ||
| Scheme 24 Mechanism of the four-component Ugi reaction. | ||
When 3-bromophenyl glyoxaldehyde 82 and N-Boc-(4,5-dimethyl)-1,2-DAB 5k were used instead of amine and aldehyde components in the above reaction the process proceeded smoothly in methanol at ambient temperature resulting in the Ugi adduct 84 in a 70% yield (Scheme 25). The treatment of 84 with TFA provides 85a in good yield, which is the first reported synthesis of quinoxalines derived in one step with the use of the Ugi reaction, assuming to proceed unexpectedly through intermediates 86 and 87 with the elimination of a benzoyl group (Scheme 26).82 The final product is appreciably different from the Ugi precursor, which provides a unique opportunity to achieve such a difference.
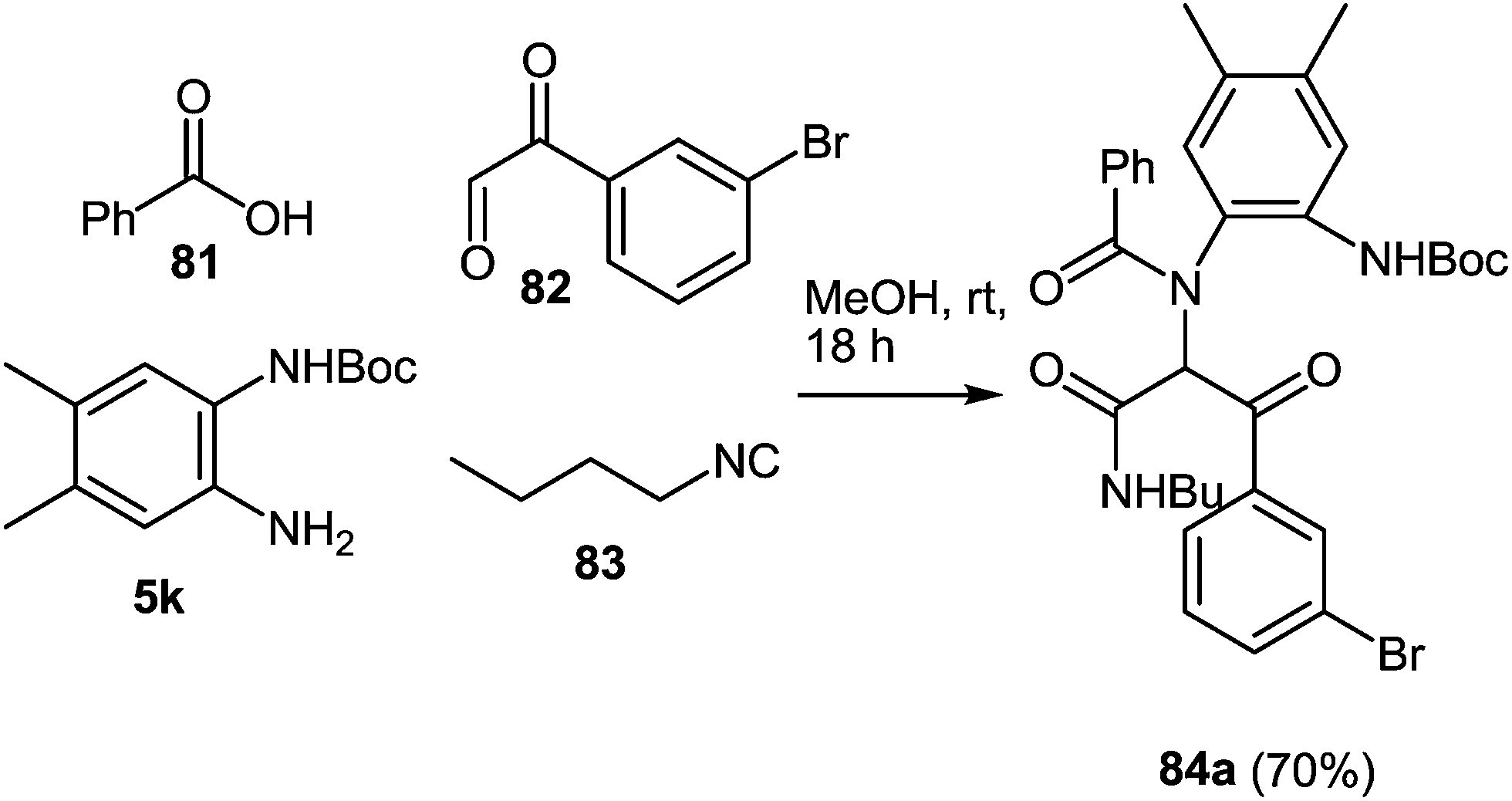 | ||
| Scheme 25 Four-component one-pot Ugi reaction. | ||
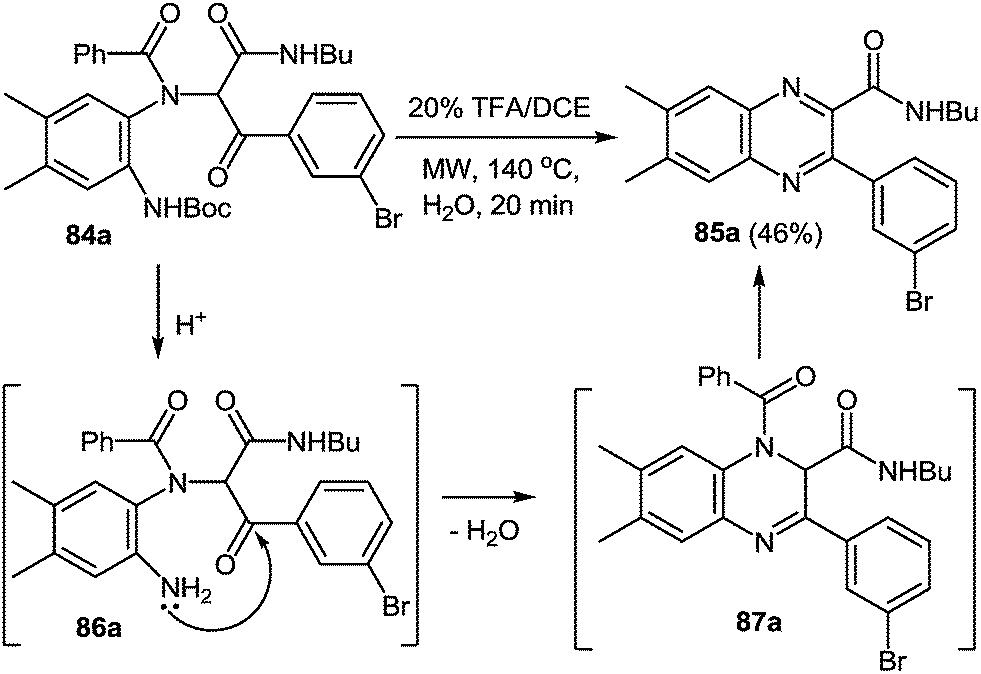 | ||
| Scheme 26 Conversion of Ugi adduct into quinoxaline 85avia intermediates 86a and 87a. | ||
One pot procedure results in a higher overall yield of 85a (46%) when compared to that of the two step process (34%) (Scheme 26).87
As to the scope of the reaction, various aldehydes, 1,2-DABs and isonitriles were used to produce a set of diversified quinoxalines and, the transformation worked equally well for all inputs (Scheme 27).87
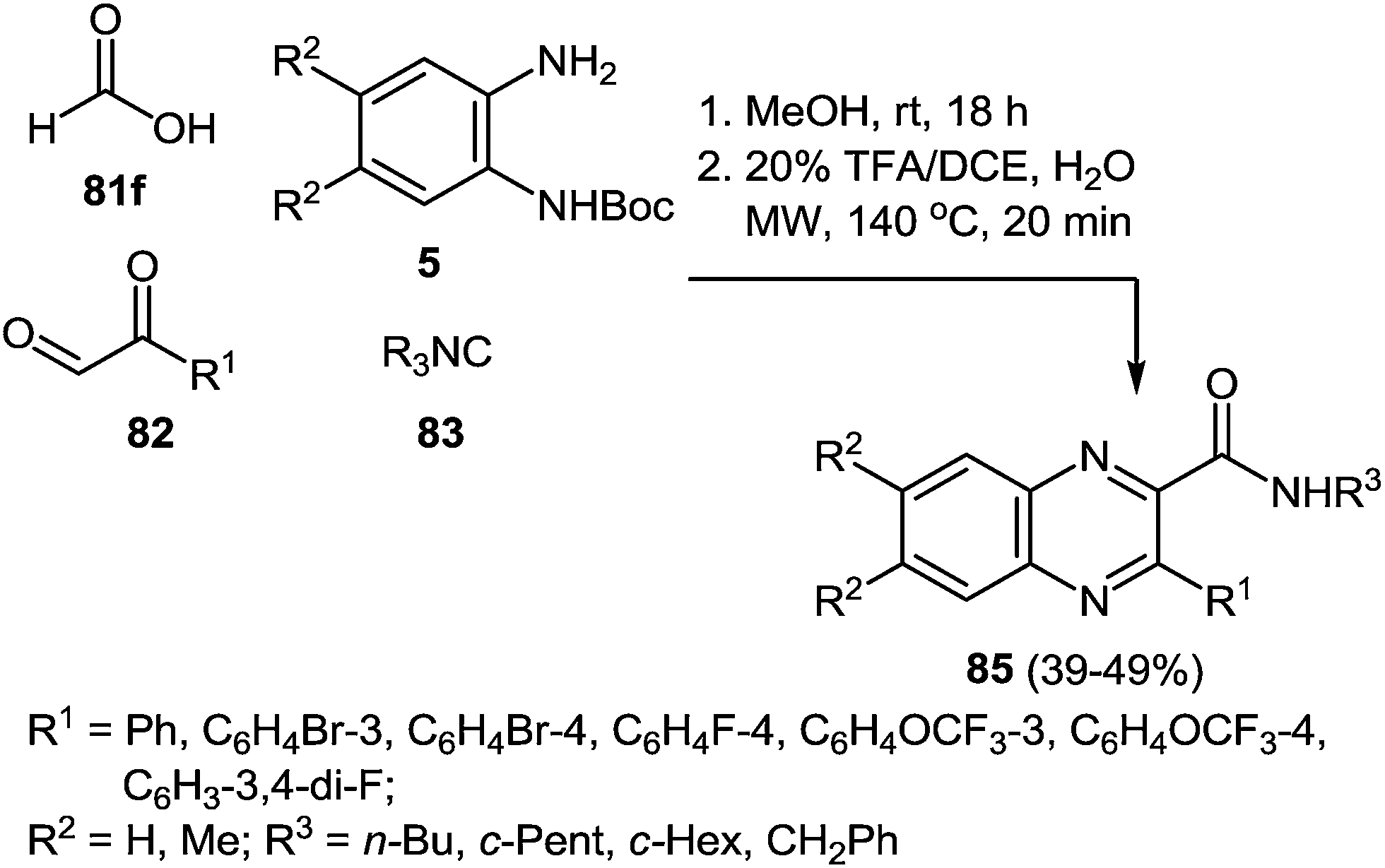 | ||
| Scheme 27 Study of the reaction scope. | ||
In the same reaction conditions, it was assumed that the use of isonitrile 86 (o-N-Boc-phenylisonitrile)88 usually facilitates access to 2-benzimidazolylquinoxalines 88.
The Ugi reaction proceeded smoothly and upon microwave irradiation of Ugi products 87 with a 20% TFA/DCE, 2-benzimidazolylquinoxalines 88a–c were obtained in good yield (Scheme 28).87 2-Benzimidazolylquinoxaline products 89 were observed with bulky o-substituted aromatic acids which had retained the benzoyl group through an internal acyl-transfer from the nitrogen obtained from the original Ugi amine input to the adjacent benzimidazole. Low yields of the desired products (39–49% in the cases of quinoxaline derivatives, and 37–46% in the case of 2-benzimidazolylquinoxalines) and the limited availability of mono- and di-variously-substituted derivatives of o-N-Boc-phenylisonitrile 86 and N-Boc-1,2-DAB limits the possibility of this approach to the synthesis of bicyclic derivatives of quinoxalines and 2-benzimidazolylquinoxalines variously substituted by quinoxaline and benzimidazole.
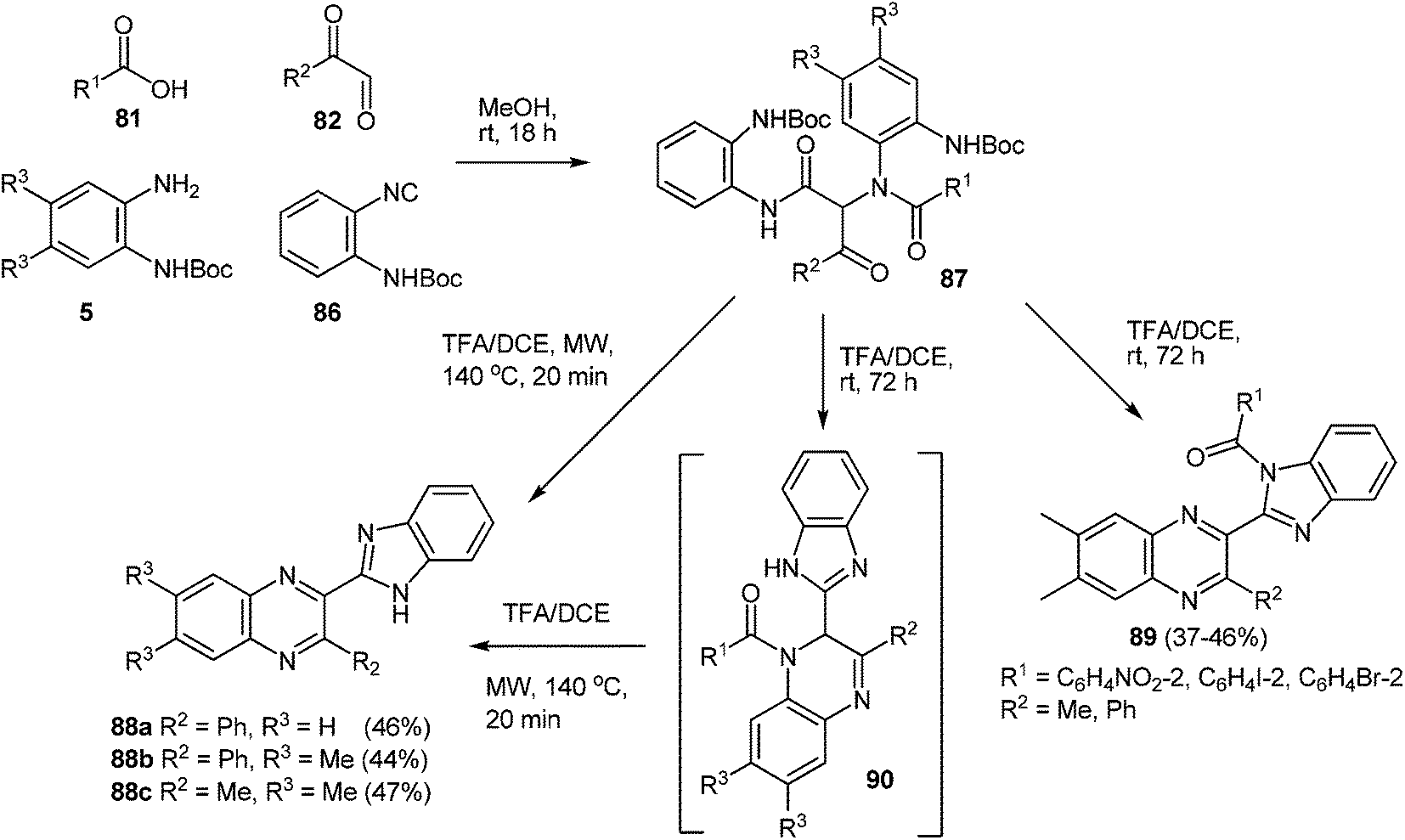 | ||
| Scheme 28 Synthesis of 2-benzimidazolylquinoxalines. | ||
4 Synthesis of 2-hetarylquinoxalines via rearrangements
4.1 Synthesis of 2-(benzimidazol-2-yl)quinoxalines and their aza-analogues
One of the common reactions of α-diketones in the chemistry of heterocycles is the Hinsberg reaction,65i.e., the synthesis of quinoxalines by the interaction of 1,2-DABs with α-dicarbonyl compounds.Thus a series of reactions according to Hinsberg were carried out.
The reaction of quinoxalin-2-one 37a (see Subsection 2.2) with 1,2-DAB 5a in boiling acetic acid leads to the corresponding 2-benzimidazolylquinoxaline 40a in a 97% yield (Scheme 29).62a
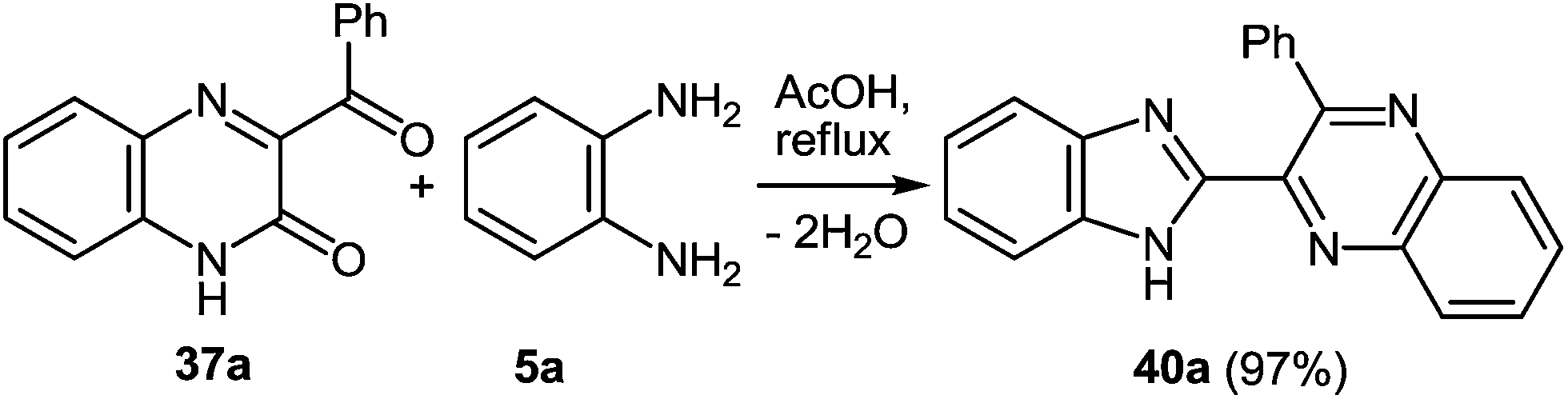 | ||
| Scheme 29 Rearrangement of 3-benzoylquinoxalin-2(1H)-one. | ||
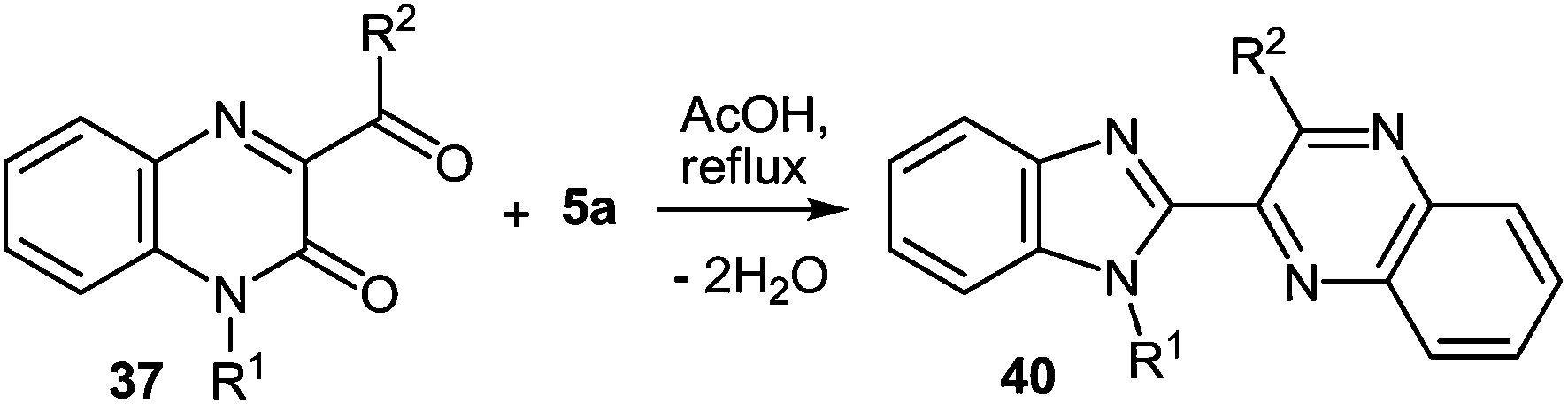
Tables 1 and 2 show that a variety of quinoxalinones 37a–r and 1,2-DABs 5a, d, h are compatible with these reaction conditions, with diverse 2-benzimidazolyl substituted quinoxalines in good yields. The reactions of 3-phenylacetylquinoxalin-2(1H)-one 37g with 3,4-diaminotoluene 5d, or 4-nitro-1,2-DAB 5h, produce a mixture of two isomers in almost equal amounts (Table 2),62d,89 as evident from the 1H NMR spectra of the crude products.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | Product | Yield (%) | Ref. |
| 1 | 37b | H | C6H4F-4 | 40b | 94 | 83 |
| 2 | 37c | H | C6H4Cl-4 | 40c | 97 | |
| 3 | 37d | H | C6H4Br-4 | 40d | 95 | |
| 4 | 37e | H | C6H4I-4 | 40e | 93 | |
| 5 | 37f | H | C6H4NO2-4 | 40f | 92 | |
| 6 | 37g | H | CH2Ph | 40g | 81 | 82 |
| 7 | 37h | H | CH2CH2Ph | 40h | 87 | |
| 8 | 37i | H | n-Pr | 40i | 82 | |
| 9 | 37j | H | Me | 40j | 72 | 81 |
| 10 | 37k | Et | Me | 40k | 99 | |
| 11 | 37l | Me | Ph | 40l | 81 | 79 |
| 12 | 37m | Et | Ph | 40m | 79 | |
| 13 | 37n | n-Pr | Ph | 40n | 87 | |
| 14 | 37o | n-Bu | Ph | 40o | 87 | |
| 15 | 37p | n-Pent | Ph | 40p | 86 | |
| 16 | 37q | CH2Ph | Ph | 40q | 56 | |
| 17 | 37r | COMe | Ph | 40r | 81 | |
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 37 | R1 | 5 | R2 | Products (yield) | Ref. |
| 1 | 37a | Ph | 5d | Me | 91a + 92a | 79 |
| (82%) | ||||||
| 2 | 37g | CH2Ph | 5d | Me | 91b + 92b | 82 |
| (34%) (44%) | ||||||
| 3 | 37a | Ph | 5h | NO2 | 91c + 92c | 79 |
| (39%) (44%) | ||||||
| 4 | 37b | C6H4F-4 | 5h | NO2 | 91d + 92d | 83 |
| (47%) (38%) | ||||||
| 5 | 37c | C6H4Cl-4 | 5h | NO2 | 91e + 92e | |
| (42%) (36%) | ||||||
| 6 | 37d | C6H4Br-4 | 5h | NO2 | 91f + 92f | |
| (45%) (43%) | ||||||
| 7 | 37e | C6H4I-4 | 5h | NO2 | 91g + 92g | |
| (44%) (45%) | ||||||
| 8 | 37g | CH2Ph | 5h | NO2 | 91h + 92h | 82 |
| (36%) (49%) | ||||||
This is because the fact that the probability of an initial attack of the amino group on the C(3) atom of the quinoxalin-2(1H)-one system and on the aroyl- or alkanoyl group during the rearrangement is nearly the same (Table 1).62a,b,d,e,90
Table 1, shows that it makes no deference for the reaction whether or not there is a substituent of quinoxalin-2(1H)-ones at position 1. The same process is successful with N-alkylated derivatives of quinoxalin-2(1H)-ones, producing N-alkylated derivatives of benzimidazole as well. All these reactions involve the formation of 2-(benzimidazol-2-yl)quinoxalines almost in quantitative yields.
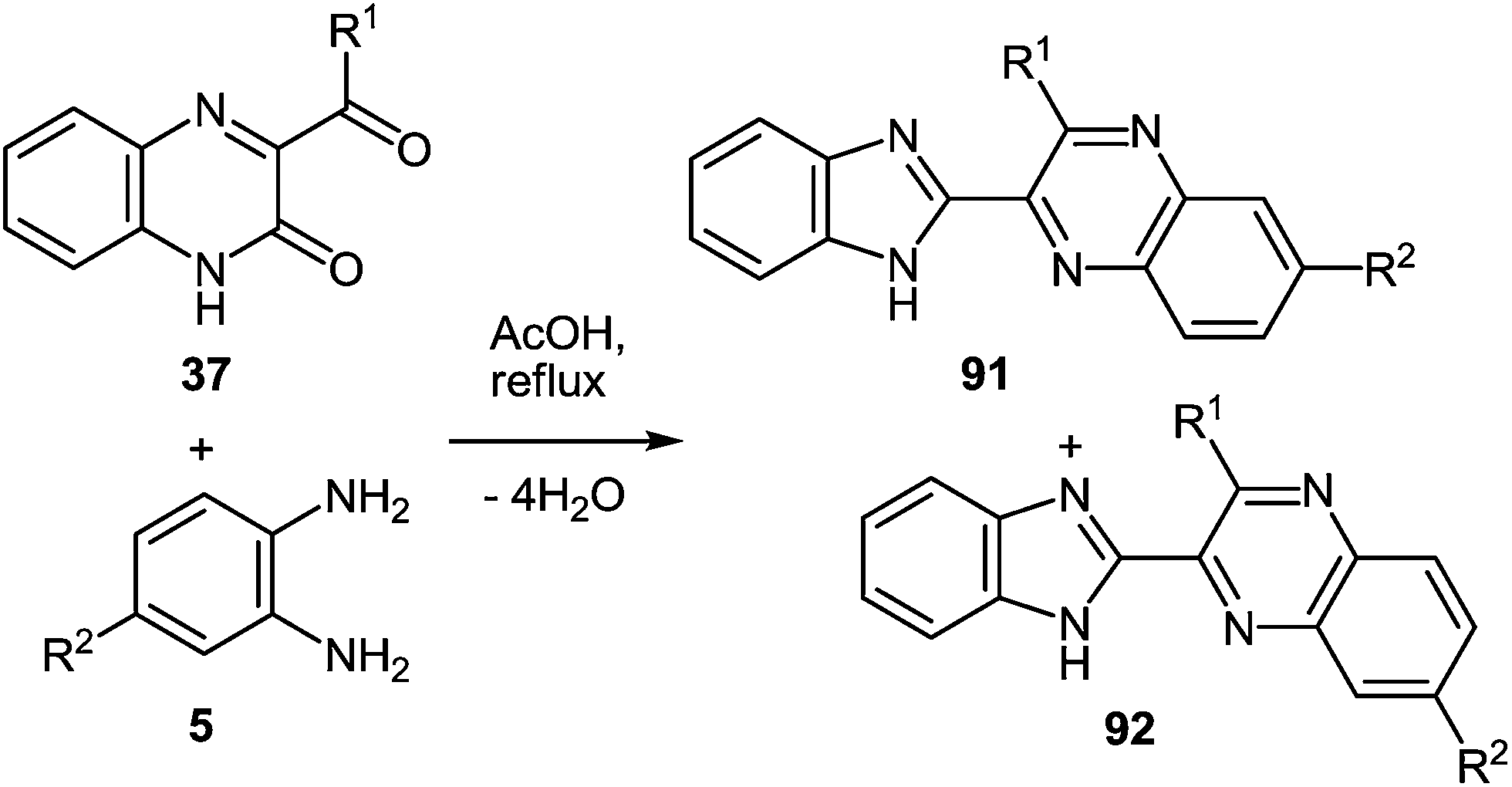
The reaction proceeds with the compounds with two quinoxalin-2(1H)-one moieties in their composition, and, what kind of spacer connects these two fragments makes no difference. In all cases the reactions proceed smoothly with the formation of benzimidazolemonopodands with the terminal quinoxaline fragments (Table 3).62c,d,91
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | X | n | Product | Yield (%) |
| 1 | 93a | O | 1 | 94a | 80 |
| 2 | 93b | O | 2 | 94b | 75 |
| 3 | 93c | CH2 | 1/3 | 94c | 69 |
| 4 | 93d | CH2 | 4/3 | 94d | 72 |
| 5 | 93e | CH2 | 8/3 | 94e | 73 |
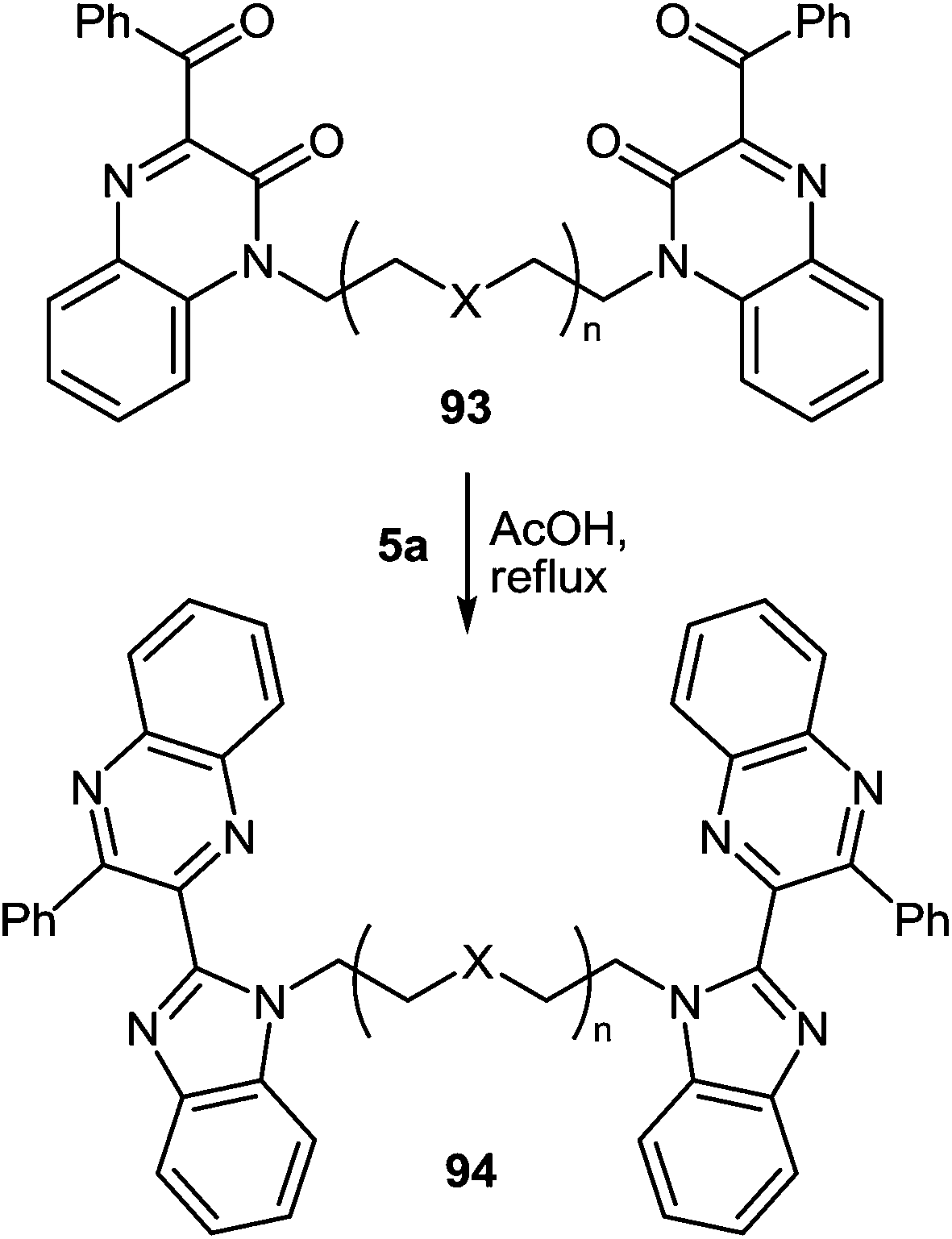
When the reaction of 1,5-bis(3-benzoyquinoxalin-2(1H)-on-1-yl)pentane 93f with the 3,4-diaminotoluene 5d was carried out under the same conditions the formation of a single product mainly the benzimidazolemonopodand 94f with the terminal 6- and 7′-dimethylquinoxaline rings in a 77% yield actually occurs (Scheme 30).62c
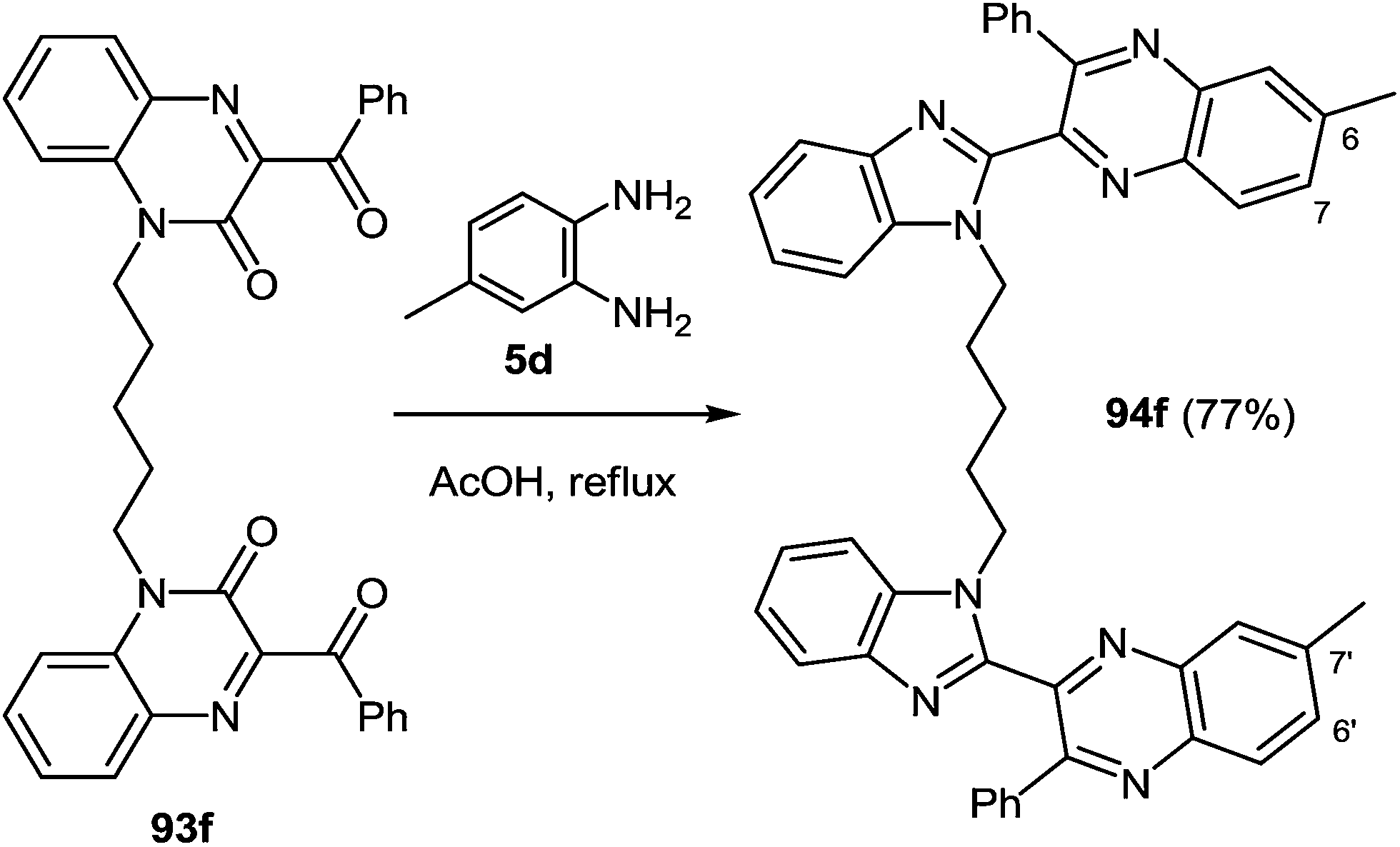 | ||
| Scheme 30 Preparation of the nonsymmetric benzimidazolemonopodand. | ||
These systems contain methyl groups at C(6) and C(7′), and not at the C(6), C(6′) and C(7), C(7′) positions. The probability of an initial attack on the amino group on the C(3) atom or on the benzoyl group is roughly the same. Otherwise, the formation of three products of rearrangement could be observed in approximately equal amounts.
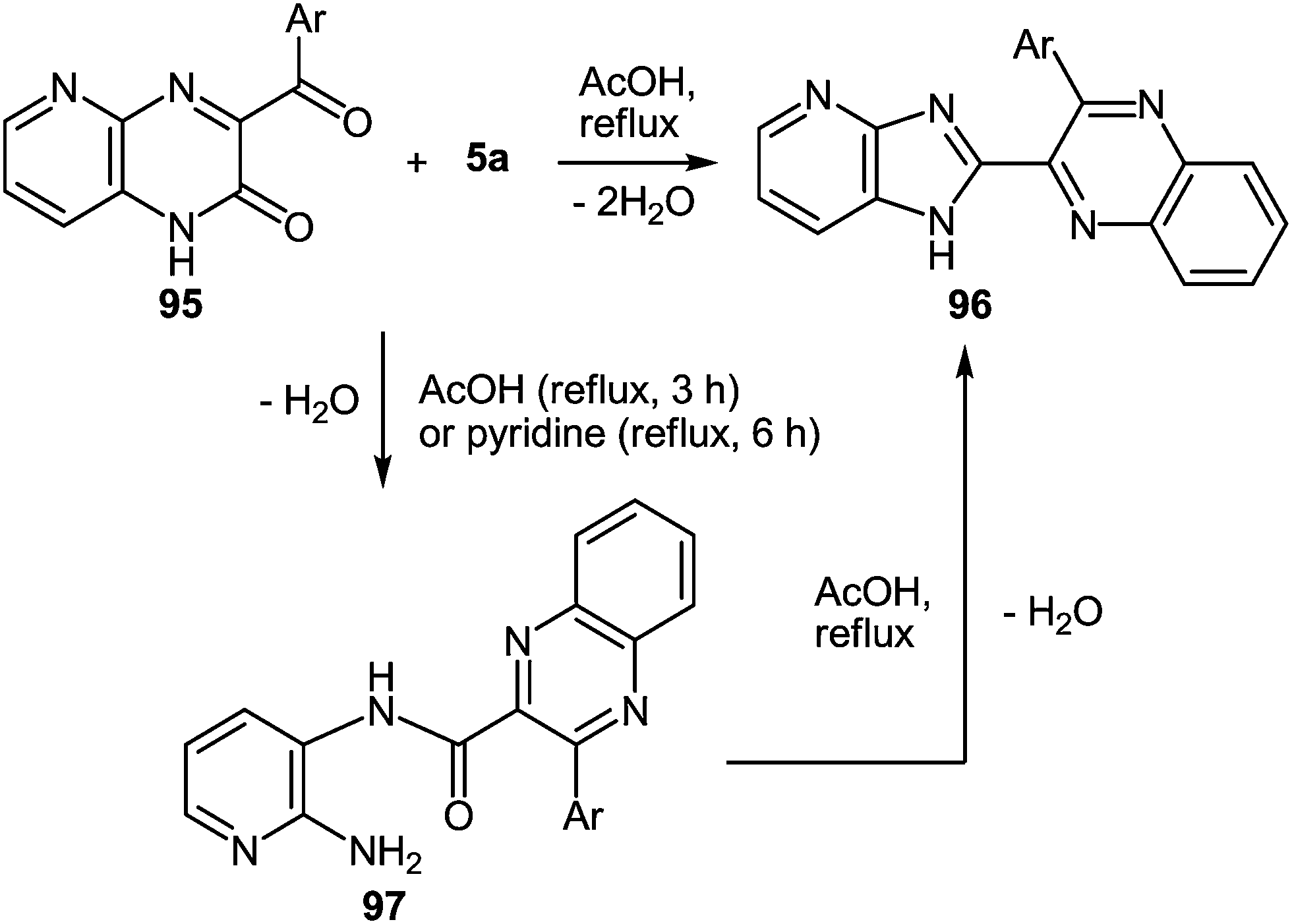
The use of aza-analogues of quinoxalinones 37 – pyrido[2,3-b]pyrazin-3(4H)-ones 95 instead of quinoxalinones 37 in the rearrangement considered above makes it possible to synthesize aza-analogues of benzimidazoles – 1H-imidazo[4,5-b]pyridines 96 which are not easily accessible by classical Phillips–Ladenburg3a,46b,47c,92b–e and Weidenhagen71,72,74,92e–g reactions. The formation of 2-(1H-imidazo[4,5-b]pyridin-2-yl)-3-arylquinoxalines 96a–d from pyrido[2,3-b]pyrazin-2(1H)-ones 95a–d and 1,2-DAB 5a proceeds in AcOH under reflux for 35–47 h. It should be noted, that the reaction with refluxed AcOH for 3 h leads to the formation of 2-amino-3-azaanylide-quinoxaline-3-phenyl-2-carboxylic acids 97. This has been illustrated by the reaction of 2-(1H-imidazo[4,5-b]pyridin-2-yl)-3-phenylquinoxaline 95a and 1,2-DAB 5a (Table 4).62f
Compounds 96a–d, in contrast to the compounds 40a–d with the benzimidazole system in the investigated solutions of DMSO-d6 exist as a tautomeric mixture of 96a–d ⇆ 96′a–d, resulting in the dissymmetric 1H-imidazo[4,5-b]pyridine system (Fig. 5). Benzimidazoles as imidazoles with a N-hydrogen ring are subjected to tautomerism, which becomes evident in unsymmetrically substituted compounds.93 In the cases of imidazo[4,5-b]pyridines 96a–d the dissymmetry was caused by the nitrogen atom of the pyridine ring.
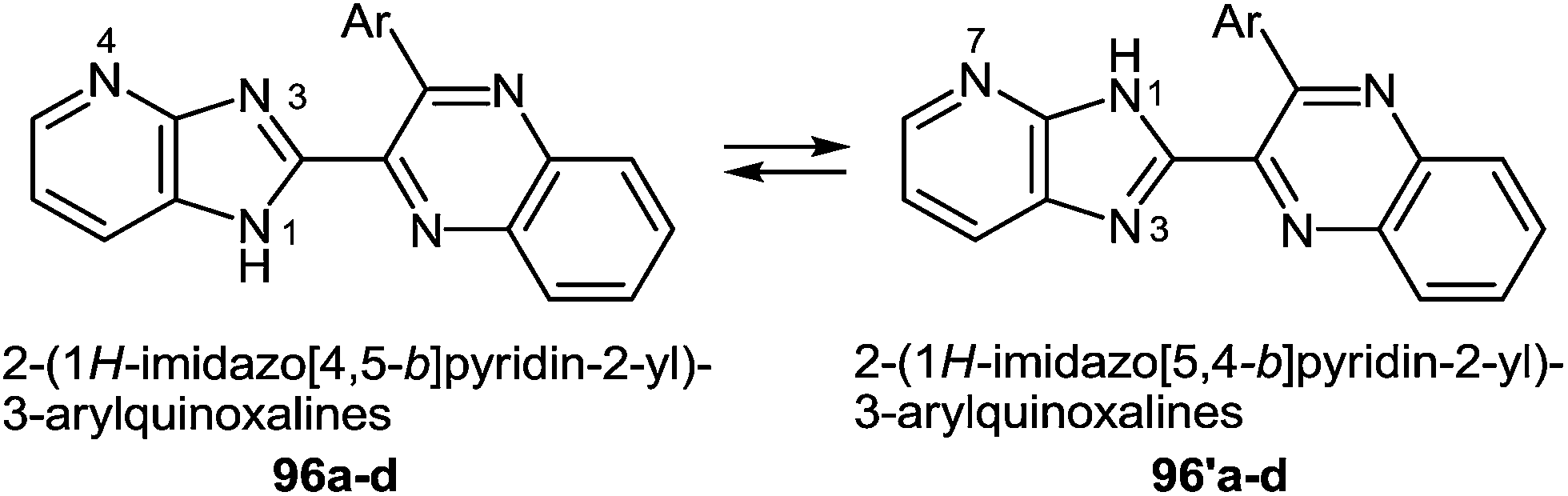 | ||
| Fig. 5 Tautomerism in imidazopyridines. | ||
4.2 Synthesis of 2,3-bis(benzimidazol-2-yl)quinoxalines
It is not a simple task to obtain high results in the synthesis of heterocyclic systems with two directly connected benzimidazole fragments with the known methods (by Phillips–Ladenburg3a,46b,47c,92b–e and Weidenhagen71,72,74,92e–g reactions). At least twice as many labor-consuming classical methods of the synthesis of benzimidazoles are involved.It should be pointed out that there was only one paper94 in which the formation of 2,3-bis(benzimidazol-2-yl)quinoxaline 100a as a by product in the reaction of tetrachloropyridazine 98 and 1,2-DAB 5a in N-methylpyrrolidone at 115 °C for 17 h has been described (Scheme 31). The yield of the main product of this reaction 5,6,7,8,13,14-hexaazapentaphene 99 obtained as a free base after treatment of the corresponding hydrochloride with aqueous sodium hydroxide is 15%. The yield of the by-product – 2,3-bis(benzimidazol-2-yl)quinoxaline 100a has not been given in this paper. Therefore this method cannot be used as a preparative one for the synthesis of 2,3-bis(benzimidazol-2-yl)quinoxalines. The synthesis of other heterocyclic systems with two benzimidazole fragments cannot be used either.
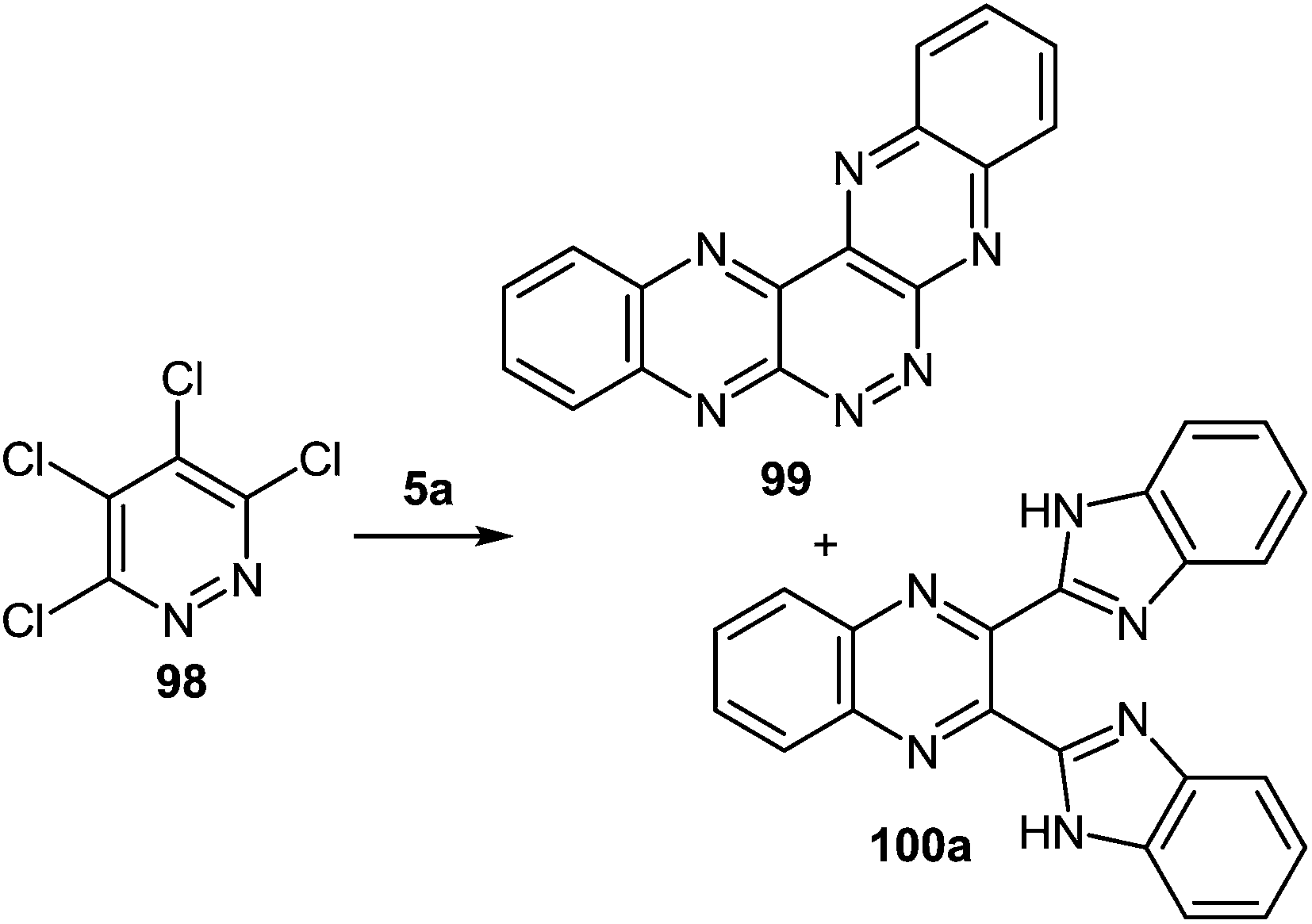 | ||
| Scheme 31 The reaction of tetrachloropyridazine and 1,2-DAB. | ||
It was suggested that the quinoxaline residue of the product 100a was formed by the initial nucleophilic attack on position 4 and 5 of tetrachloropyridazine 98 by 1,2-DAB. Further the aerial oxidation, resulting in an intermediate 1,4-dichloropyridazino[4,5-b]quinoxaline 101 which might react with two more molecules of 1,2-DAB to give the pyridazine-ring-opened system 100a (Scheme 32).
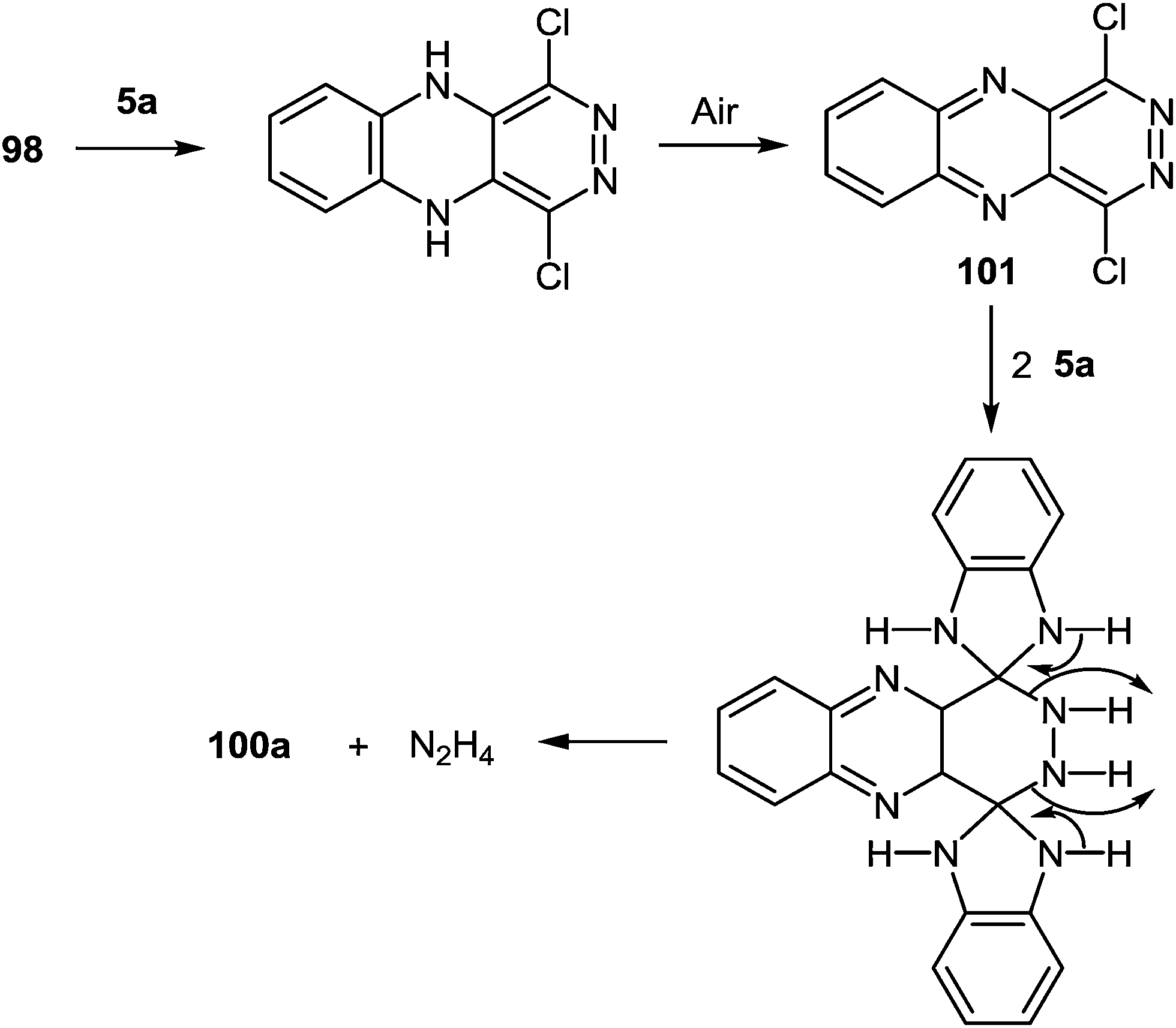 | ||
| Scheme 32 Displacement of hydrazine hydrochloride from 1,4-dichloropyridazino[4,5-b]quinoxaline by 1,2-DAB. | ||
As seen from the above data (see Section 4.1), the rearrangement proceeds very well with different aroyl- and alkanoyl-derivatives of quinoxalinones and as a result various benzimidazole derivatives are obtained. A further analysis of this strategy led us to examine how this rearrangement would proceed, if 3-hetaroylquinoxalin-2(1H)-ones were used instead of alkanoyl- or aroylquinoxalin-2(1H)-ones. Among heteroaroyl groups the benzimidazoyl group was of primary interest since on the one hand, the successful course of the reactions of the 3-(benzimidazo-2-yl)quinoxalin-2(1H)-one95 with 1,2-DABs opens up a new and effective way of obtaining 2,3-bis(benzimidazol-2-yl)quinoxaline derivatives. The latter are inaccessible by any other known methods of constructing the benzimidazole system. On the other hand, this could prove whether the interaction of 3-(benzimidazo-2-yl)quinoxalin-2(1H)-one with 1,2-DAB dihydrochloride resulted in the benzodiazepine derivative, as previously cited in literature95 (Scheme 33).
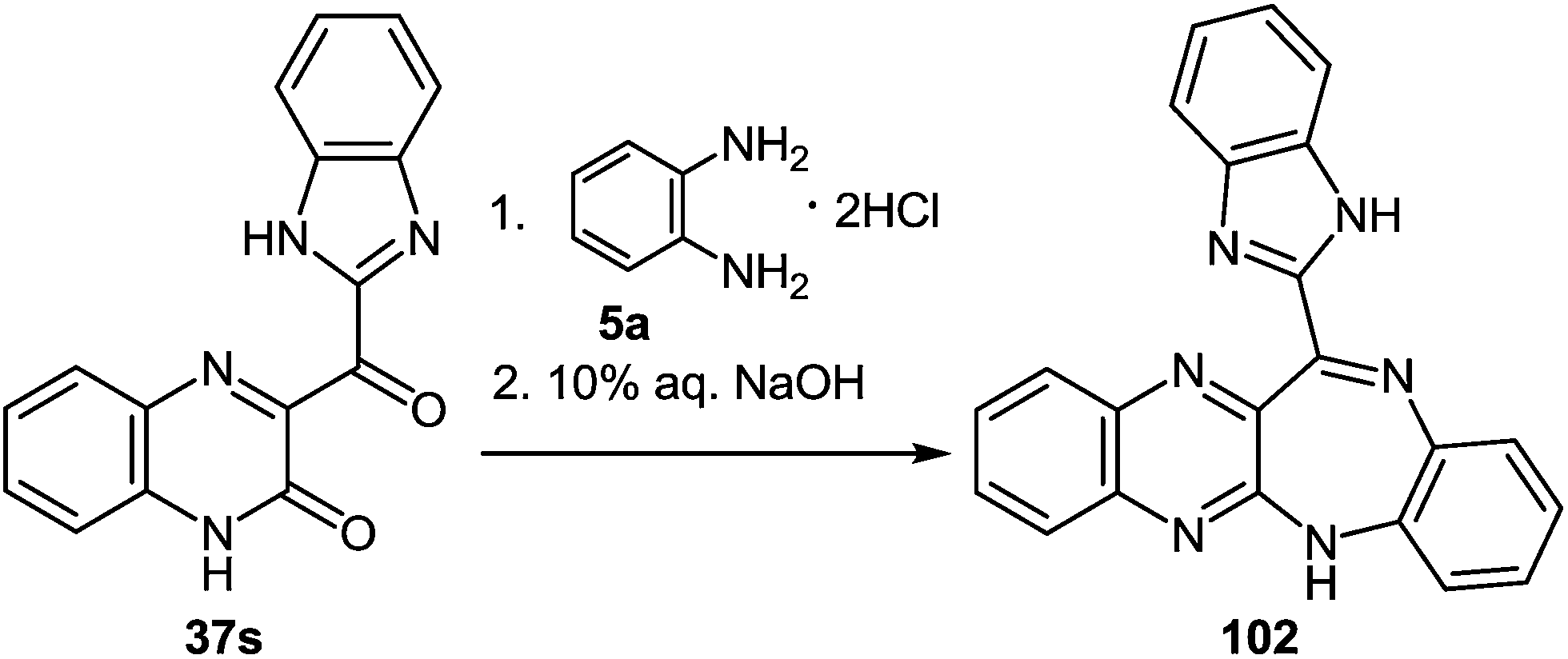 | ||
| Scheme 33 The interaction of 3-benzimidazoylquinoxalin-2(1H)-one 37s with 1,2-DAB dihydrochloride as described in literature.95 | ||
Kurasawa in his work95 showed that the reaction of 3-benzimidazoylquinoxalinone 37s with 1,2-DAB dihydrochloride in boiling acetic acid resulted in a benzodiazepine derivative 102 (Scheme 33).
Since this is contrary to the above findings on the reactions of aroylquinoxalin-2(1H)-ones with 1,2-DAB,62,96 proceeding with the formation of 2-benzimidazolylquinoxaline derivative it was decided to examine the results described in paper.95 To this end, it was planned to synthesize 3-benzimidazoylquinoxalinone according to the following Scheme 34 described by Kurasawa.95,97
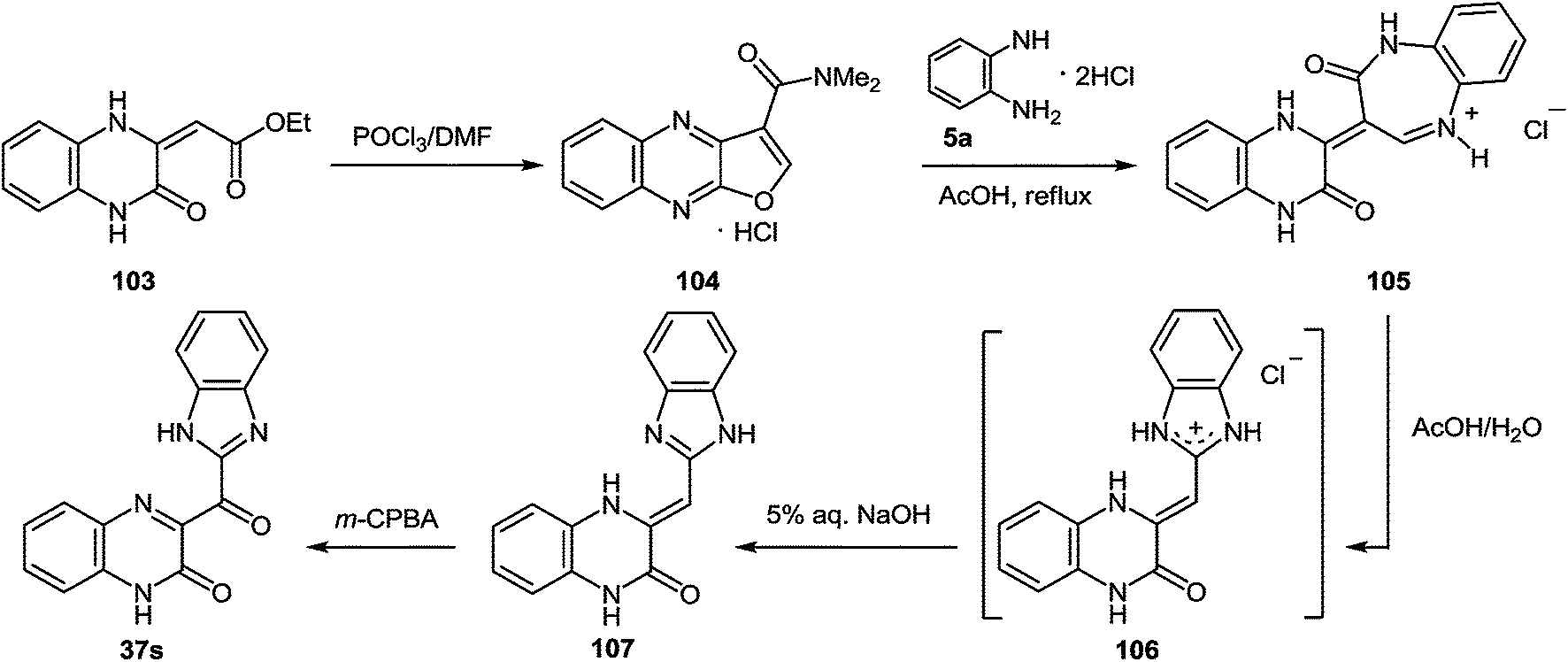 | ||
| Scheme 34 The general scheme of the synthesis of 3-benzimidazoylquinoxalin-2(1H)-one 37s starting with quinoxalin-2(1H)-one derivative 103 according to the references 95,97 The geometry of CC double bond for quinoxaline derivative 107 is strongly supported by gauge-independent atomic orbital calculations.99 | ||
As depicted in Scheme 34 compound 103 at the very first stage to react with the Vilsmeier reagent [DMF–POCl3, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1], at a temperature on water bath for 2 h, to give 3-(N,N-dimethylaminocarbonyl)furo[2,3-b]quinoxaline hydrochloride 104 (83%).97a After a detailed study of the structure of the reaction product, with the involvement of IR data98 it was concluded that there was no compound 104, but its isomer – 1-N,N-dimethylaminomethylene-1,2-dihydrofuro[2,3-b]quinoxalin-2-one 108 (Scheme 35).
:1], at a temperature on water bath for 2 h, to give 3-(N,N-dimethylaminocarbonyl)furo[2,3-b]quinoxaline hydrochloride 104 (83%).97a After a detailed study of the structure of the reaction product, with the involvement of IR data98 it was concluded that there was no compound 104, but its isomer – 1-N,N-dimethylaminomethylene-1,2-dihydrofuro[2,3-b]quinoxalin-2-one 108 (Scheme 35).
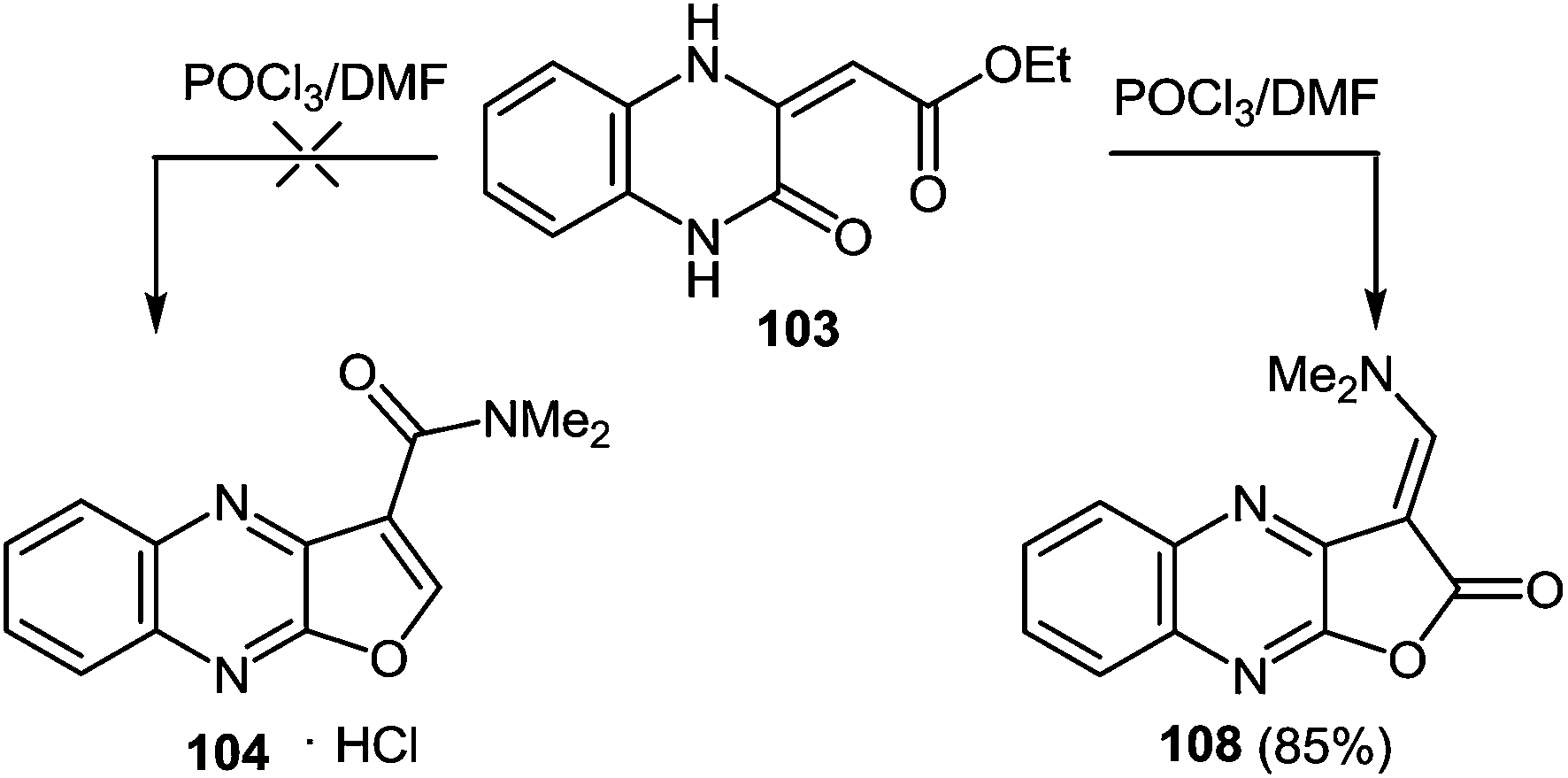 | ||
| Scheme 35 The reaction of 3-ethoxycarbonylmethylene-3,4-dihydroquinoxalin-2(1H)-one 103 with the Vilsmeier reagent. | ||
Apparently the formation of the furo[2,3-b]quinoxalin-2-one derivative 108 proceeds via the initial formation of compound A according to the first stage of the aliphatic Vilsmeier reaction, performed on the methylene group of the substituent at position 3 of quinoxalin-2(1H)-one 103. The latter undergoes intramolecular cyclization with the subsequent elimination of EtOH and HCl, results in the final product 108 (Scheme 36).
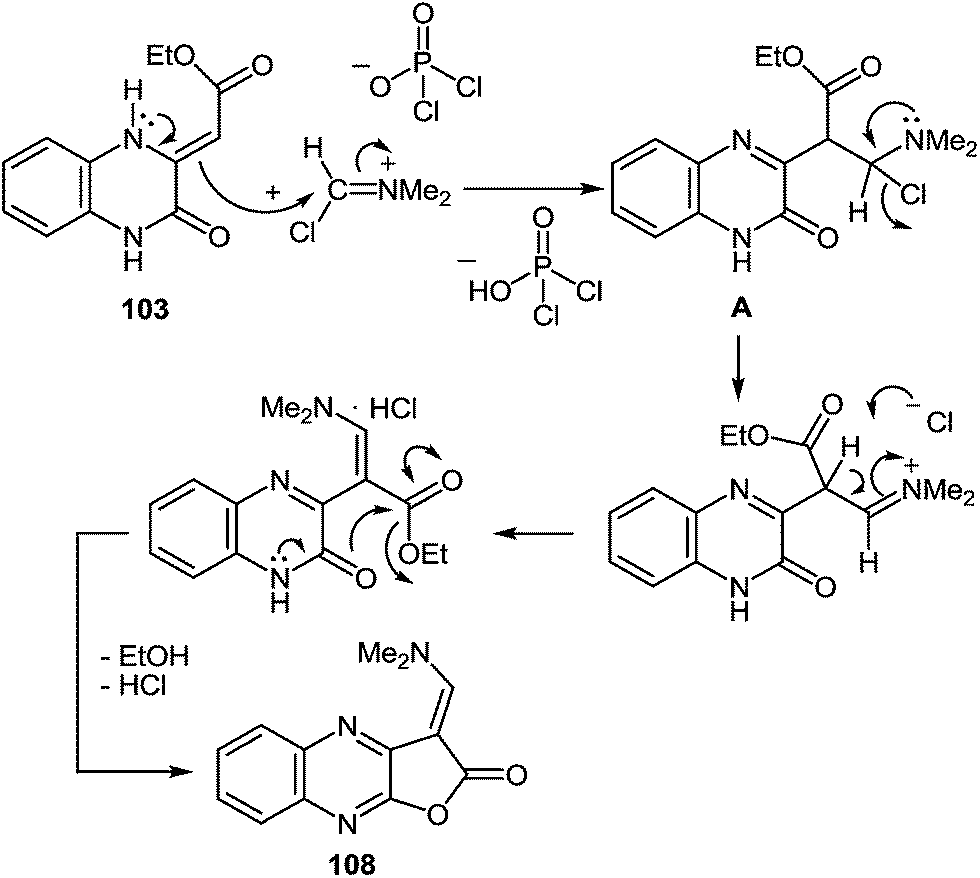 | ||
| Scheme 36 A plausible mechanism for the formation of furo[2,3-b]quinoxalin-2-one derivative 108. | ||
The next stage of the synthesis of 3-benzimidazoylquinoxalin-2(1H)-one 37s according to Scheme 34 is the reaction of 104 with 1,2-DAB dihydrochloride in acetic acid which resulted in the ring transformation to afford 3-(3,4-dihydroquinoxalin-2(1H)-on-3-yl)-1,2-dihydro-1,5-benzodiazepin-2(1H)-one hydrochloride 105.97b
The reaction of 108 with 1,2-DAB dihydrochloride in the same conditions afforded a compound, which was identified by NMR spectroscopy as 3-(3,4-dihydroquinoxalin-2(1H)-on-3-yl)-1,2-dihydro-1,5-benzodiazepin-2(1H)-onium chloride 105.97b The formation of benzodiazepin-2(1H)-one derivative 105 can be due to the attack of the amino group of 1,2-DAB dihydrochloride on the activated carbon atom of the N,N-dimethylaminomethylene group of furo[2,3-b]quinoxalin-2-one derivative 108 by the HCl and results in the formation of intermediate B. The latter undergoes intramolecular ring closure and ring opening processes, by the addition of the nitrogen atom of the second amino group of 1,2-DAB to the carbonyl group of the furan ring. The cleavage of the C–O bond results in the intermediate C. The 1,5-benzodiazepine system C is subsequently transformed into the final product 105 as shown above (Scheme 37).
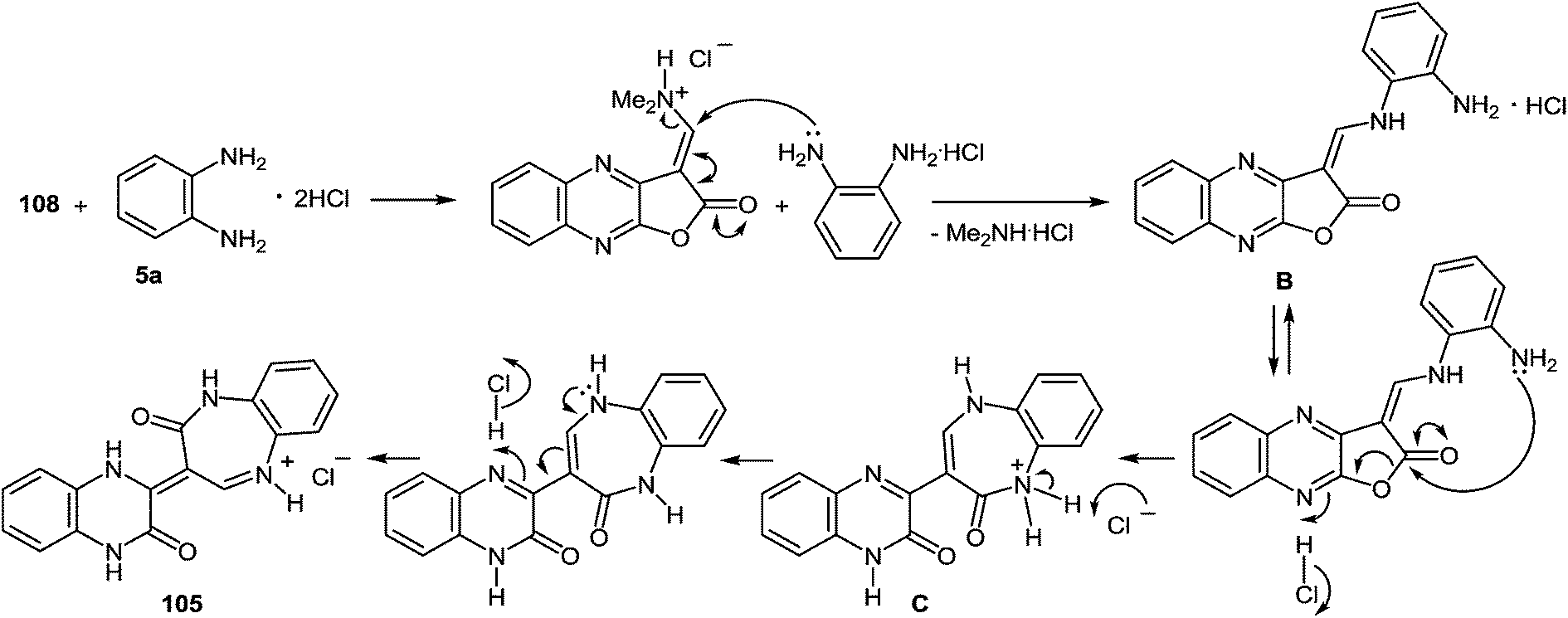 | ||
| Scheme 37 A possible mechanism of the formation of benzodiazepin-2(1H)-one derivative 105. | ||
A possible mechanism for the formation of benzodiazepin-2(1H)-one derivative 105 (Scheme 38) can be alternatively written as follows; the amination with 1,2-DAB dihydrochloride would occur at the carbonyl carbon of 108, forming the corresponding amide of 1,2-DAB and the subsequent intramolecular Michael addition of mono-N-acylated 1,2-DAB D to form the 1,5-benzodiazepine system C, subsequently transformed into the final product 105 (Scheme 38).
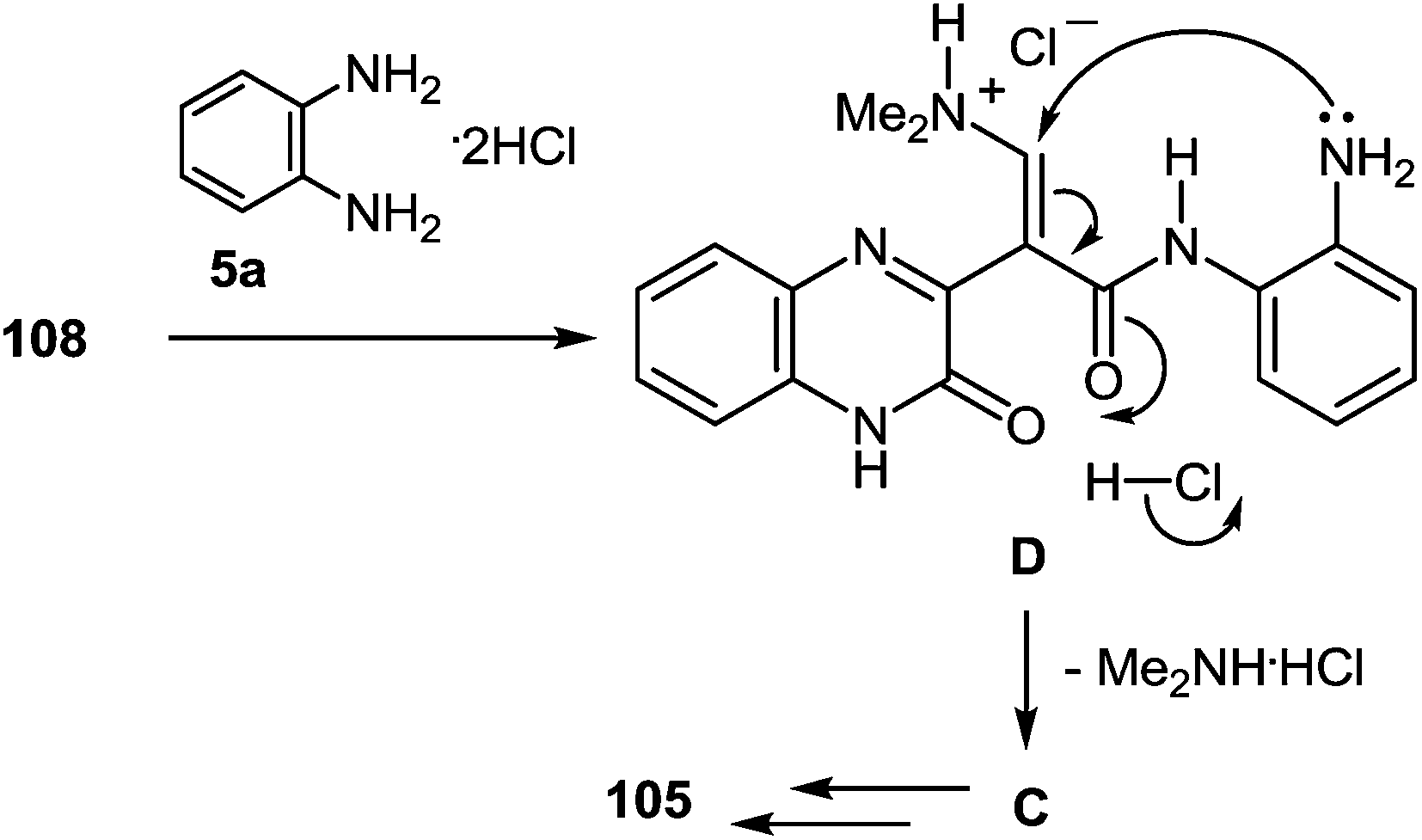 | ||
| Scheme 38 A possible alternative mechanism for the formation of benzodiazepin-2(1H)-one derivative 105. | ||
Heating at reflux of the benzodiazepine derivative 105 in the aqueous acetic acid solution affected ring transformation as has been shown in paper95 to give 106, the treatment of which with a 5% sodium hydroxide yielded a free base 107 (Scheme 34). The final stage of the synthesis of 3-benzimidazoylquinoxalin-2(1H)-one 37s according to Scheme 34 involves the oxidation of compound 107 with m-chloroperbenzoic acid (m-CPBA).95 According to this method the yield of the desired product is approximately 40%, therefore it was used the method of oxidation for the synthesis of 3-benzimidazoylquinoxalin-2(1H)-one 37s, which has been recently developed for similar compounds. This includes the treatment of 3-(α-bromobenzyl)quinoxalin-2(1H)-ones with DMSO.63b The reaction of 3-(benzimidazol-2-yl)methylenequinoxalin-2(1H)-one 107 with an equimolar amount of bromine in DMSO at room temperature for 24 h results in ketone 37s with a quantitative yield. According to the Kornblum oxidation100 the formation of the latter can be represented as an oxo-dehydrohalo-bisubstitution101 formed in situ 3-(α-bromomethylimidazol-2-yl)quinoxalin-2-(1H)-one (Scheme 39).
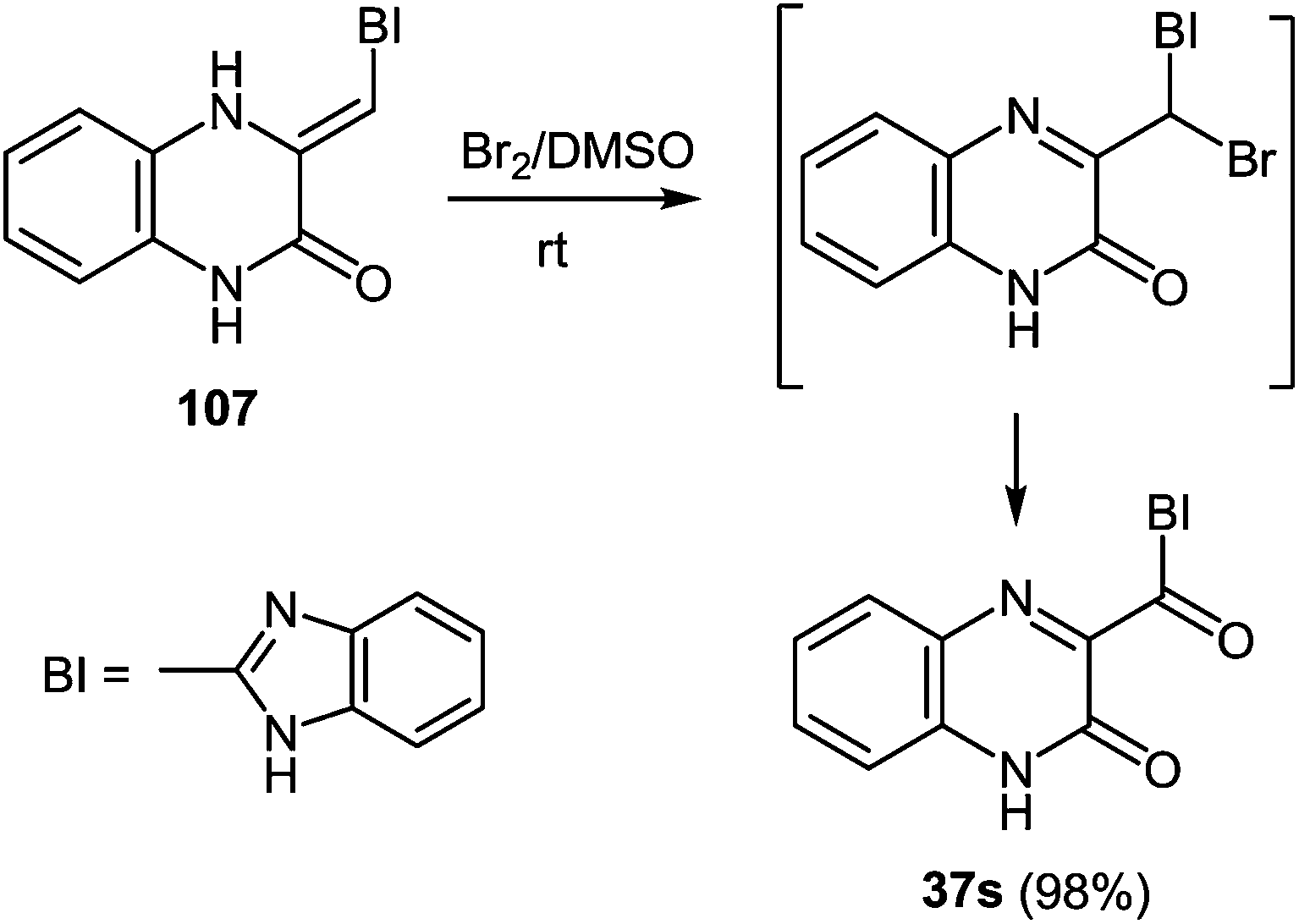 | ||
| Scheme 39 The synthesis of 3-benzimidazoylquinoxalin-2(1H)-one 37s. | ||
As expected the reaction of 3-(benzimidazo-2-yl)quinoxalin-2(1H)-one 37s with 1,2-DAB dihydrochloride, proceeds with the formation of 2,3-bis-(1H-benzimidazol-2-yl)quinoxaline 100a (Scheme 40), but not of the benzodiazepine derivative 102, as described.95 In this reaction when 1,2-DAB is used instead of 1,2-DAB dihydrochloride the yield of compound 100a is almost quantitative.
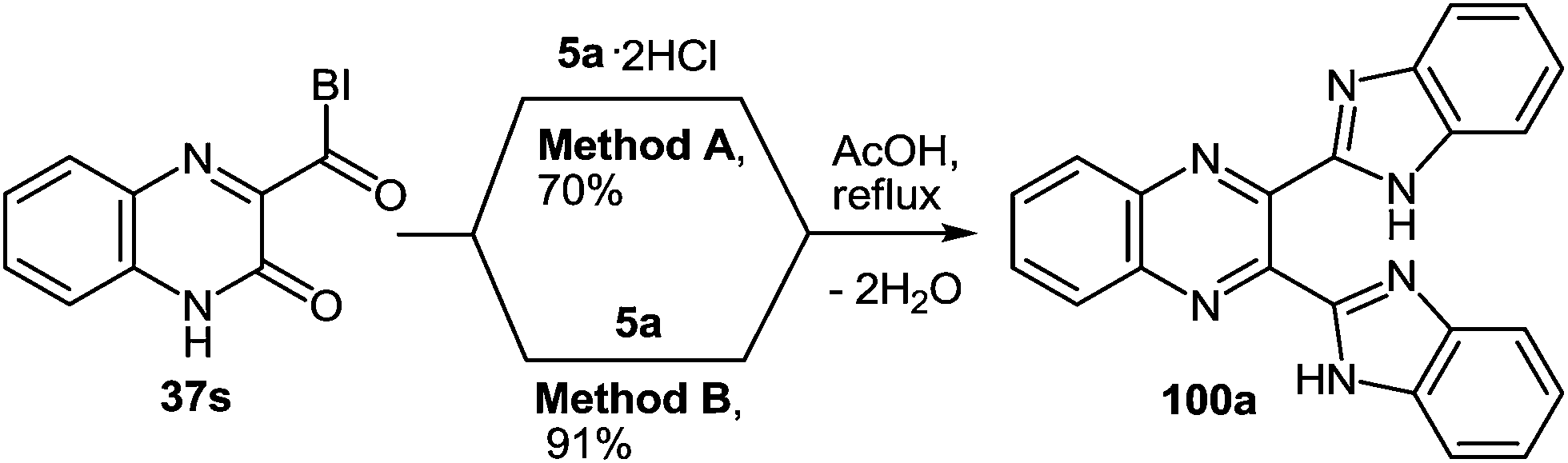 | ||
| Scheme 40 The quinoxalinone-benzimidazole rearrangement in the reaction of 3-(benzimidazo-2-yl)quinoxalin-2(1H)-one 37s with both 1,2-DAB dihydrochloride and 1,2-DAB. | ||
Thus, the reactions of 3-benzimidazoylquinoxalin-2(1H)-one 37s with both 1,2-DAB dihydrochloride and 1,2-DAB proceed according to the new quinoxalinone-benzimidazole rearrangement. There occurs the formation of 2,3-bis-(1H-benzimidazol-2-yl)quinoxaline 100a and not of the benzodiazepine derivative 102, as has been previously described. The reactions of the synthesis of 3-benzimidazoylquinoxalin-2(1H)-one 37s starting from 3-ethoxycarbonylmethylene-3,4-dihydroquinoxalin-2(1H)-one through the 3-(3,4-dihydroquinoxalin-2(1H)-on-3-yl)-1,2-dihydro-1,5-benzodiazepin-2(1H)-one hydrochloride 105 and 3-(benzimidazol-2-yl)methylenequinoxalin-2(1H)-one 107 have also been reexamined and the published results95,97 appropriately amended. A simple and efficient one-pot method for the synthesis of 3-benzimidazoylquinoxalin-2(1H)-one 37s directly from 3-(benzimidazol-2-yl)methylenequinoxalin-2(1H)-one 107 has been described.
3-(Benzimidazo-2-yl)quinoxalin-2(1H)-one 37s reacted not only with 1,2-DAB 5a and its substituted analogues 5c, d, e, h, l, m in AcOH at reflux. The formation of 2,3-bis(benzimidazol-2-yl)quinoxalines 100b–g was observed in excellent 76–90% isolated yields (Table 5).86
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | Product | Yield (%) |
| 1 | 5c | Me | Me | 100b | 90 |
| 2 | 5d | Me | H | 100c | 86 |
| 3 | 5f | Cl | H | 100d | 89 |
| 4 | 5h | NO2 | H | 100e | 83 |
| 5 | 5l | CO2H | H | 100f | 76 |
| 6 | 5m | COPh | H | 100g | 80 |
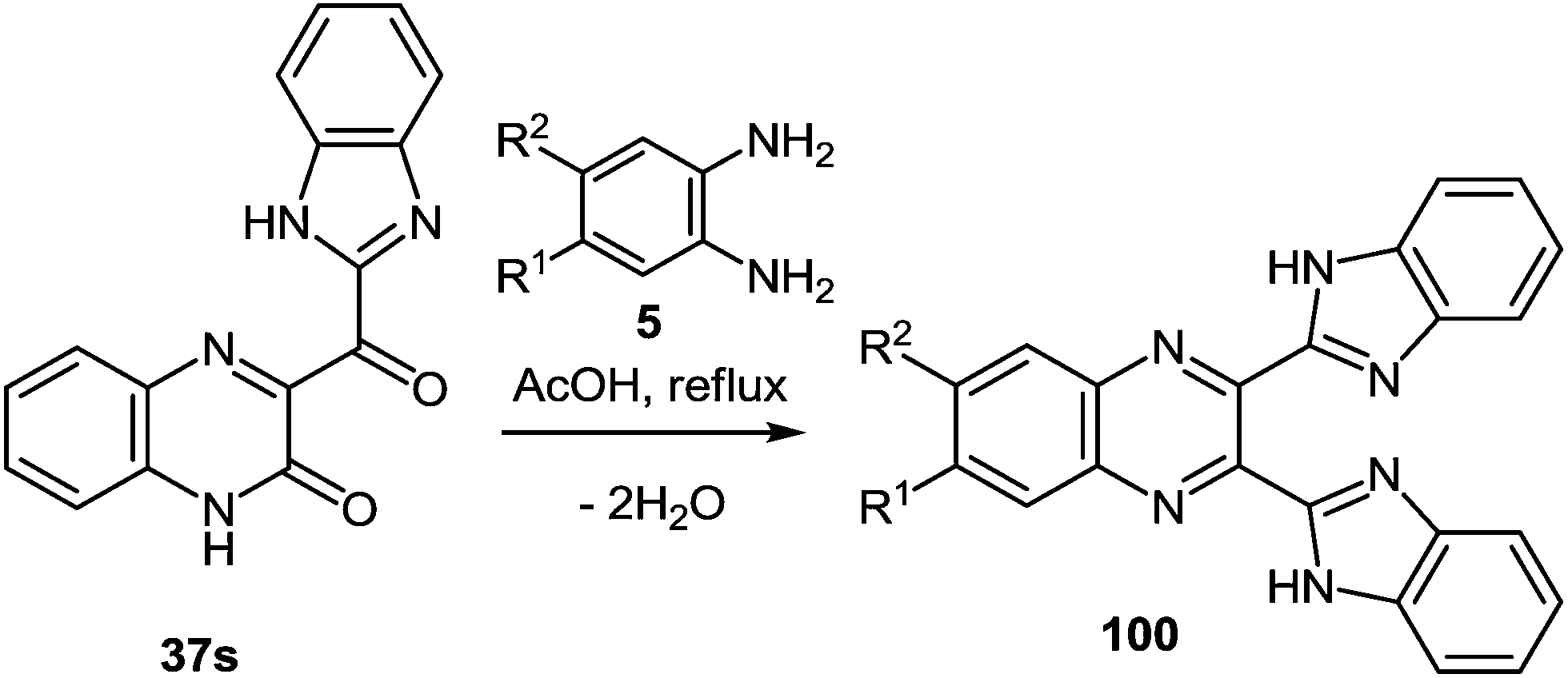
The scope of the methodology, was studied first with respect to the 3,3′-diaminobenzidine 113 (Scheme 41). As seen, this chemistry is not limited to mono- and disubstituted systems, a compound with two 1,2-diaminobenzene fragments is an acceptable substrate as well.102
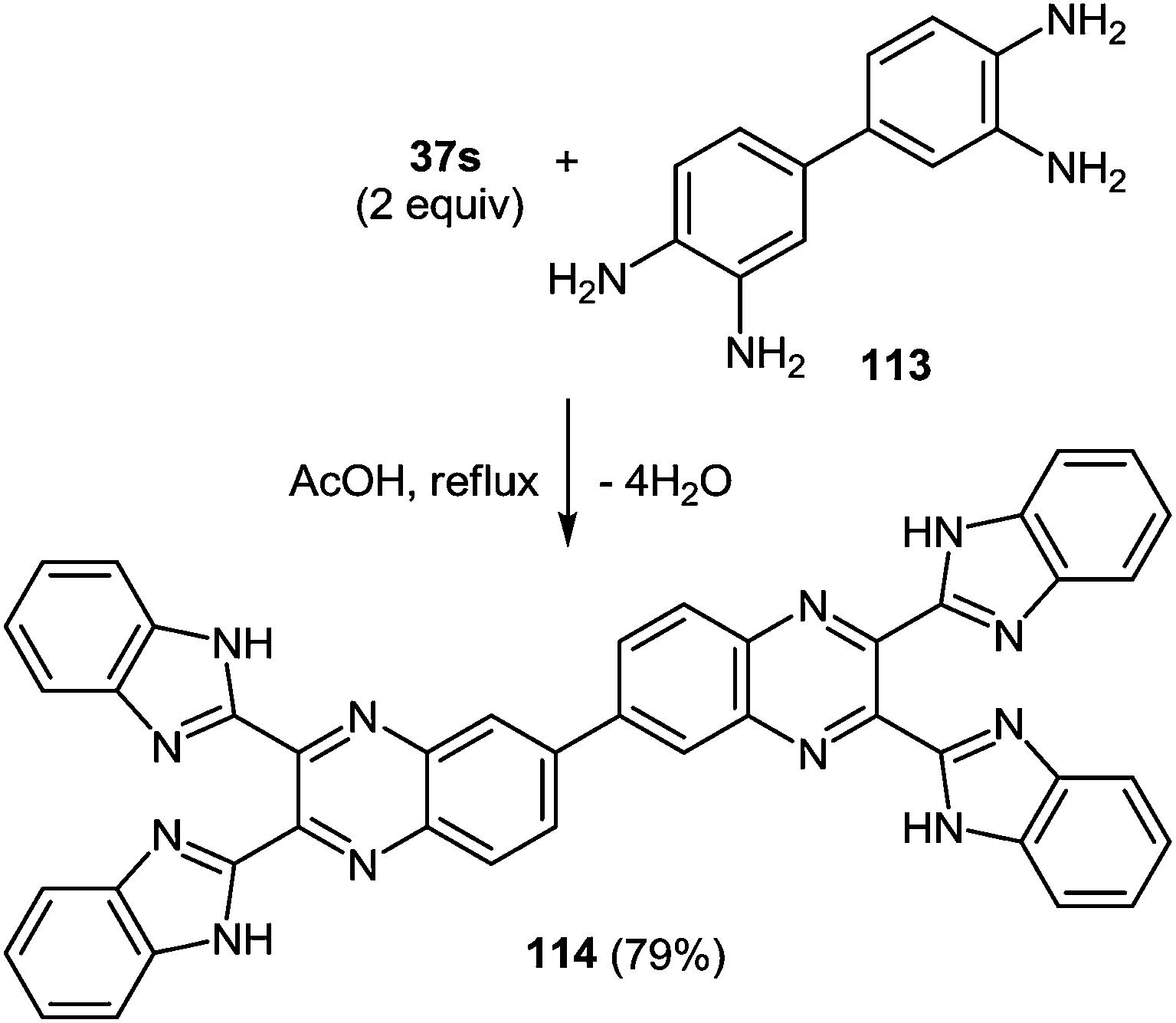 | ||
| Scheme 41 The formation of 2,2′,3,3′-tetra(benzimidazol-2-yl)-6,6′-biquinoxaline. | ||
Investigations were also carried out with respect to the condensed 1,2-DABs. The 3-(benzimidazo-2-yl)quinoxalin-2(1H)-one 37s was allowed to react with benzo[c]furane-4,5-diamine 115, benzo[c]thiene-4,5-diamine 116 and quinoxaline-5,6-diamine 117 in AcOH at reflux (Scheme 42).102 The isolated yields of the di(benzimidazol-2-yl)quinoxalines 118–120 proved high enough as well.
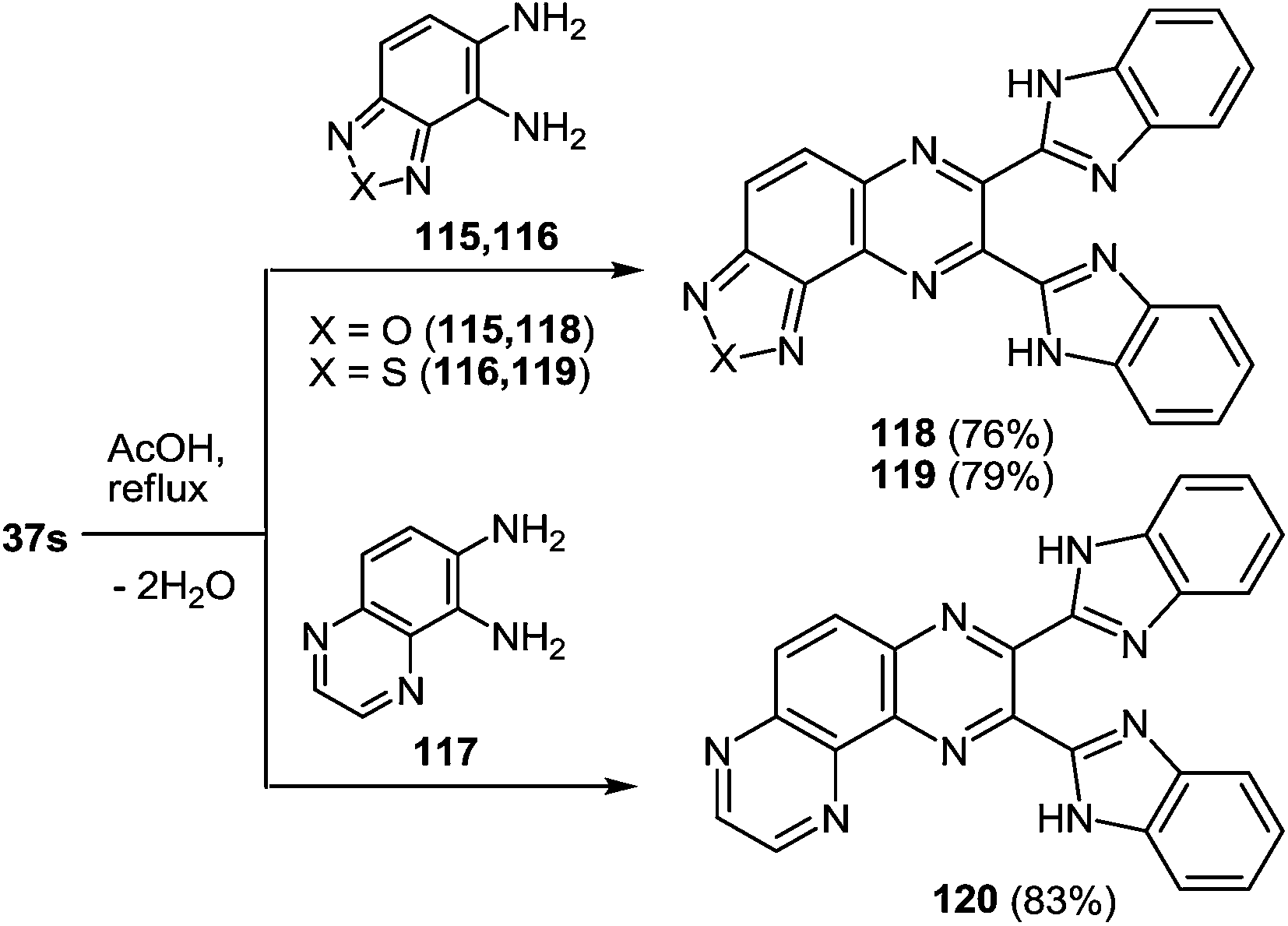 | ||
| Scheme 42 The quinoxalinone-benzimidazole rearrangement in the reaction of 3-(benzimidazo-2-yl)quinoxalin-2(1H)-one 37s with benzo[c]furane-4,5 – 115, benzo[c]thiene-4,5 – 116 and quinoxaline-5,6 – 117 diamines. | ||
Thus, the results of the reaction of 3-(benzimidazo-2-yl)quinoxalin-2(1H)-one 37s with a variety of 1,4-di-N-nucleophiles confirm the hypothesis, which has been proposed earlier (Scheme 16).76
Thus, we have developed a simple, mild and high yielding one-pot method to prepare quinoxalines and tethered quinoxaline-benzimidazoles. In this case the scope of the rearrangement was extended to other 1,4-di-N-nucleophiles, which enables a simple and efficient method for the preparation of a series of 2,3-bis(benzimidazol-2-yl)quinoxalines as well as of di(benzimidazol-2-yl)-furo[3,4-f]-, thieno[3,4-f]- and pyrazino[2,3-f]quinoxalines. This was accomplished by the rearrangement of 3-benzimidazoylquinoxalin-2(1H)-one on exposure to 1,2-DAB and its substituted and condensed derivatives. Considering the well documented medicinal utility of benzimidazoles and quinoxalines, these compounds consisting from the combination of the two scaffolds afford new opportunities to investigate their biological activity.
4.3 Synthesis of 2-(pyrazin-2-yl)benzimidazoles
Further exploration of the above strategy with the use of diaminomaleonitrile instead of 1,2-DABs has led to the development of a novel, simple method for the synthesis of 2-benzimidazolylpyrazines. In this case the results of study on a novel rearrangement of 3-aroyl- and alkanoylquinoxalin-2(1H)-ones and 2,3-diaminomaleonitrile as a N-nucleophile under the acid catalysis condition are presented.103Regardless of the molar ratio of the reagents (1:1, 1:1.1, 1:2; 37a:121) and reaction time (3, 9 or 17 h) the reaction of 3-benzoylquinoxalin-2(1H)-one (3-BQ) 37a with 2,3-diaminomaleonitrile 121 proceeded in the same way with the formation of a ∼45% yield of the rearrangement product 122a (Scheme 43), whereas 50% of 3-benzoylquinoxalin-2(1H)-one 37a reverted. Diaminomaleonitrile 121 apparently undergoes polymerization. Adding a second equivalent of diaminomaleonitrile 121 to the reaction mixture obtained after boiling the equimolar ratio of reagents in acetic acid for 5 h did not lead to an increased yield of the desired product. Neither did the boiling for an additional 5 h.
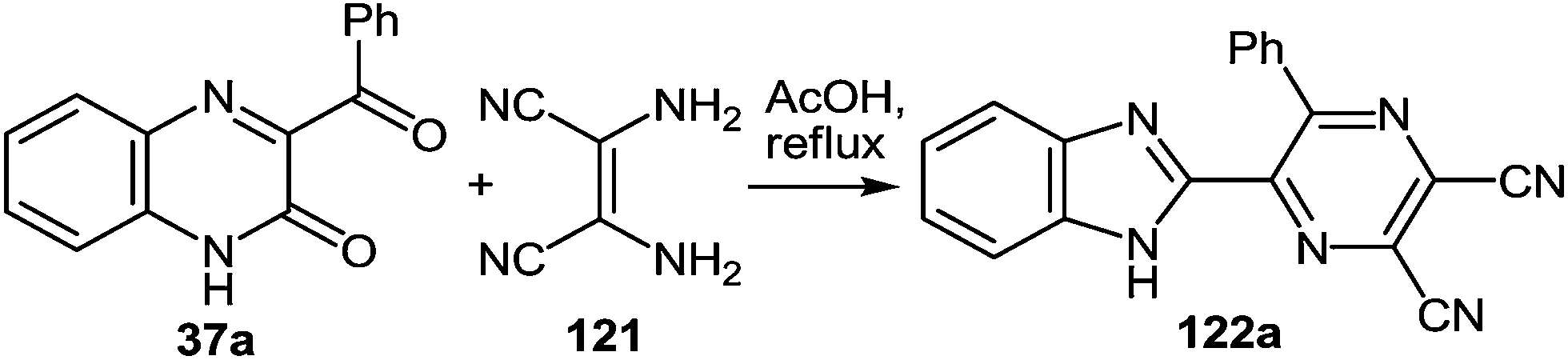 | ||
| Scheme 43 Acid-catalyzed rearrangement. | ||
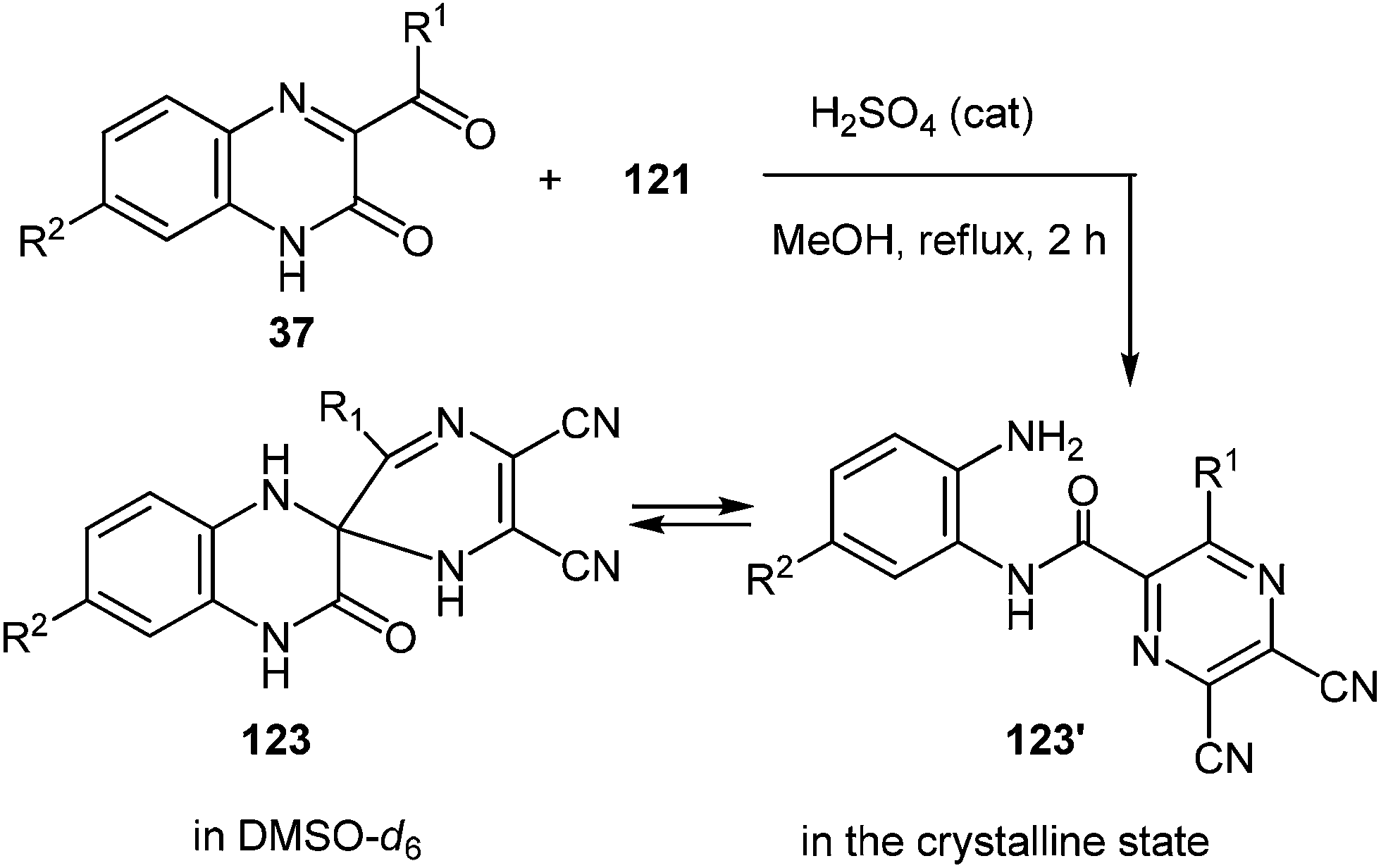
In order to improve the yield of the rearrangement product there were used the principles of the previously proposed hypothesis (Scheme 16).76 In accordance with this hypothesis here appeared the problem of the synthesis of the spiro[pyrazine-2,2′-quinoxalin]-3′(4′H)-one derivative. For this purpose the necessary pyrazine ring system at position 2 of quinoxalin-2(1H)-one 37avia the modified Körner and Hinsberg104 reaction (the synthesis of quinoxalines by condensation of α-dicarbonyl compounds with 1,2-DAB) has been installed. This was achieved by the reaction of 37a with diaminomaleonitrile 121 in a boiling solution of MeOH in the presence of a catalytic amount of H2SO4. In this reaction the 3-BQ 37a was considered as an heteroanalogue of an α-dicarbonyl compound. The reaction proceeded smoothly for 2 h to give the desired spiro-compound, 5,6-dicyano-3R-1H,1′H-spiro[pyrazine-2,2′-quinoxalin]-3′(4′H)-one 123a in 96% yield (Table 6, entry 1). The optimum molar ratio of reagents 37a:121 was 1.0:1.1. Under these conditions other 3-aroyl- (37a–e, t, u) and 3-alkanoyl- (37g, i) derivatives of quinoxalin-2(1H)-ones behaved similarly resulting in high (75–96%) yields of the corresponding spiro-derivatives on reaction with diaminomaleonitrile 121 (Table 6). These examples of the reactions of 37a, d with 121 showed that increasing the reaction time up to 10 h resulted in a mixture of the products of rearrangement of 122a, d and spiro-derivatives of quinoxalinones 123a, d in a ratio of ∼1:3 (yield 89%), and up to 20 h in a ratio of ∼1:1 (yield 97%). When carrying out the reactions of quinoxalines 37a–e, g, j, t, u with diaminomaleonitrile 121 for 6 h in the presence of p-TsOH (20 mol%) as a catalyst the formation of spiro-compounds 123a–i occurred in yields of 60–85% (Table 6). The reactions of quinoxalin-2(1H)-ones 37a, b with 121 show that in the presence of catalytic amounts of HCl, spiro-compounds 123a–b are formed in 2 h and the yields of products were 91% and 88%, respectively (Table 6, entries 1 and 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | Product | Yield (%) |
| a p-TsOH was used instead of H2SO4. b HCl was used instead of H2SO4. | |||||
| 1 | 37a | Ph | H | 123a | 96, 74a, 91b |
| 2 | 37b | C6H4F-4 | H | 123b | 92, 85a, 88b |
| 3 | 37c | C6H4Cl-4 | H | 123c | 93, 82a |
| 4 | 37d | C6H4Br-4 | H | 123d | 93, 77a |
| 5 | 37e | C6H4I-4 | H | 123e | 95, 77a |
| 6 | 37t | Ph | COPh | 123f | 95, 61a |
| 7 | 37u | Ph | CO2H | 123g | 89, 64a |
| 8 | 37g | CH2Ph | H | 123h | 95, 60a |
| 9 | 37j | Me | H | 123i | 75, 61a |
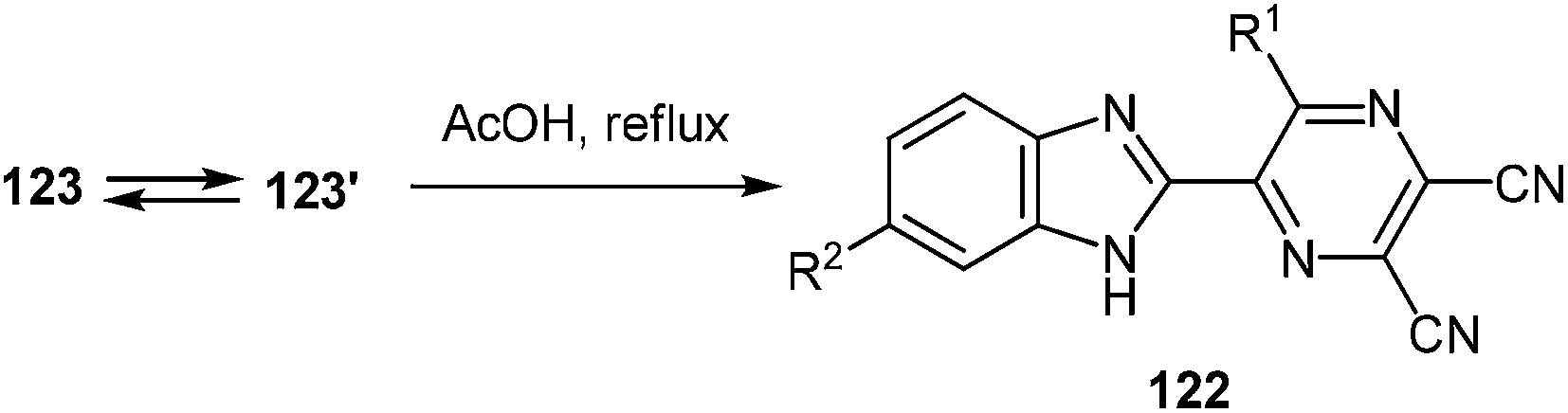
Spiro-compounds 123a–e, h, i without substituents on the benzene ring of the quinoxaline system were quantitatively transformed into the desired 2-(pyrazin-2-yl)benzimidazole 122a–e, h, i in boiling AcOH in 10 min (Table 7, entries 1–5, 8, 9). The substituted spiro-derivatives of quinoxalin-2(1H)-ones 123f, g underwent the rearrangement only after 3 h in boiling AcOH (Table 7, entries 6, 7).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Time | Product | Yield (%) |
| 1 | Ph | H | 10 min | 122a | 90 |
| 2 | C6H4F-4 | H | 10 min | 122b | 90 |
| 3 | C6H4Cl-4 | H | 10 min | 122c | 91 |
| 4 | C6H4Br-4 | H | 10 min | 122d | 89 |
| 5 | C6H4I-4 | H | 10 min | 122e | 92 |
| 6 | Ph | COPh | 3 h | 122f | 89 |
| 7 | Ph | CO2H | 3 h | 122g | 85 |
| 8 | CH2Ph | H | 10 min | 122h | 92 |
| 9 | Me | H | 10 min | 122i | 93 |
The formation of spiro-compounds also proceeded with the N-alkylated derivatives of quinoxalin-2(1H)-ones 37o, v, w to give N-alkylated spiro[pyrazine-2,2′-quinoxalin]-3′(4′H)-ones 123j–l (Table 8). p-TsOH was more suitable as the catalyst for these cases. It should be pointed out that the rearrangement of N-alkylated spiro-compounds 123j–l was slower than with nonalkylated spiro-compounds 123a–i. Thus, boiling the spiro-compounds 123j–l for 5 min resulted in the formation of the product of rearrangement in a ∼25% yield, 30 min: ∼50%, 3 h: ∼75%, 7 h: ∼100%.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R | Product | Yield (%) | Entry | R | Product | Yield (%) |
| 1 | n-Bu | 123j | 95 | 4 | n-Bu | 122j | 58 |
| 2 | n-Oct | 123k | 94 | 5 | n-Oct | 122k | 60 |
| 3 | n-Non | 123l | 97 | 6 | n-Non | 122l | 60 |
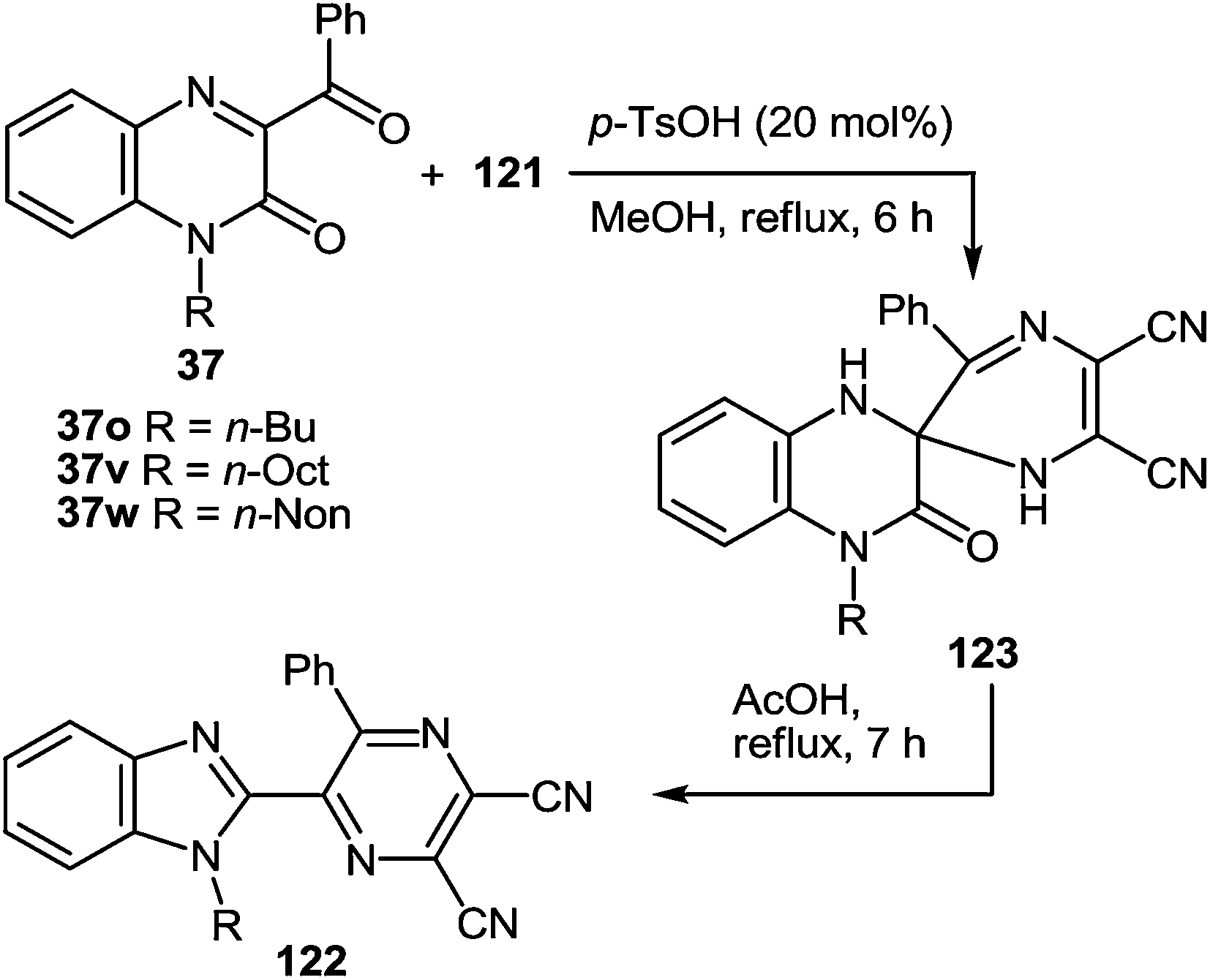
It is worthy of note that the products of the reaction of 3-aroyl(alkanoyl)quinoxalin-2(1H)-ones 37a–e, g, i, t, u and N-alkyl-3-BQs 37o, v, w with diaminomaleonitrile 121 in DMSO-d6 exist solely in spiro-cyclic-forms 123a–l, whereas in the crystalline state they exist only as open chain forms 123′a–l.103
Based of the chemistry of quinoxalinones,105 diaminomaleonitrile,106 and the above data, it is reasonable to assume that as the first step the formation of 2-(pyrazin-2-yl)benzimidazoles 122 involves addition of the amino group of diaminomaleonitrile 121 to the C(3) atom of quinoxalin-2(1H)-one 37. The next step involves a nucleophilic attack of the second amino group of 121 on the benzoyl carbonyl group to form the spiro-quinoxaline derivative 123. Rearrangement of the spiro-quinoxalinone 123 is then assumed to occur according to Scheme 46, which proceeds via cascade reactions involving: (a) acid-catalyzed ring-opening with cleavage of the C(3)–N(4) bond in the spiro-compound 123 with the formation of an intermediate quinoxaline derivative 123′, (b) intramolecular nucleophilic attack by the amino group on the carbonyl group with the formation of intermediate hydroxy-derivative A, and (c) elimination of water leading to the final product 122 (Scheme 44).
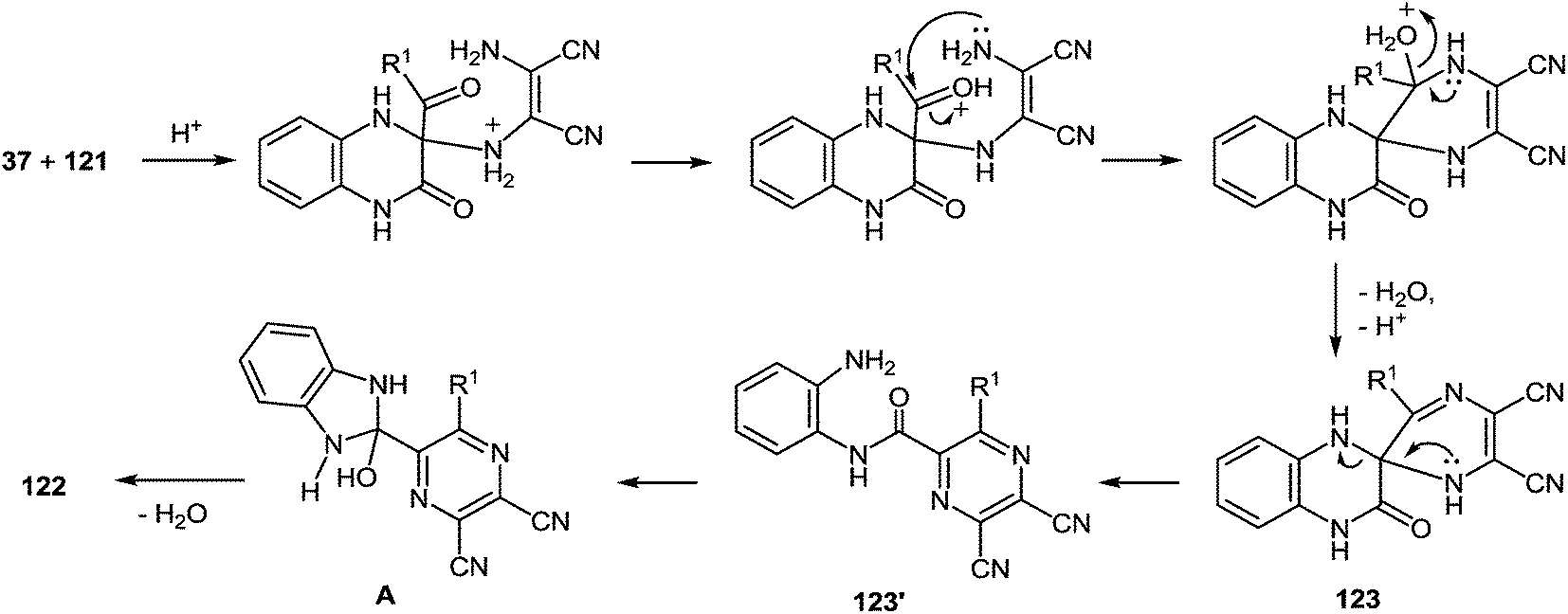 | ||
| Scheme 44 A plausible mechanism for the formation of 2-(pyrazin-2-yl)benzimidazoles 122. | ||
An efficient and versatile metal-free method for the preparation of a series of 2-(pyrazin-2-yl)benzimidazoles has been developed. This was accomplished by the novel rearrangement of 3R-5,6-dicyano-1H,1′H-spiro[pyrazine-2,2′-quinoxalin]-3′(4′H)-ones easily obtained from 3-aroyl- and 3-alkanoylquinoxalin-2(1H)-ones on exposure to diaminomaleonitrile. The key advantages are the simplicity of the operation, high yields, easy availability of 3-aroyl- and alkanoylquinoxalin-2(1H)-ones, as well as the simple work-up and purification of the products.
4.4 Synthesis of 2-(imidazol-4-yl)benzimidazoles
In continuation of the efforts to develop quinoxalin-2(1H)-one.62c,f,107 based on synthetic methodologies, a simple, mild and expeditious synthesis of 2,4,5-trisubstituted imidazoles in high yields with 3-aroylquinoxalin-2(1H)-ones as hetero analogues of α-diketones has been developed (Table 9).108|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrates | Ratio 1/2/NH4OAc | Time (h) | Products | Ratio 127/128a | Total yield (%) | |
| 127 + 128 | |||||||
| a The ratio of compounds 127 and 128 was determined by 1H NMR spectroscopy. b Isolated yield after reflux for 15 h. c Isolated yield when the reaction was carried out in the presence of L-proline. d Isolated yield of 128d = 42%. | |||||||
| 1 | 37a | 126a | 1/1/2 | 9 | 127a + 128a | 2/1 | 5 (7)b |
| 2 | 37a | 126a | 1/1/10 | 9 | 127a + 128a | 1.8/1 | 30 |
| 3 | 37a | 126a | 1/2/10 | 7 | 127a + 128a | 2/1 | 79 (79)c |
| 4 | 37a | 126a | 1/3/10 | 7 | 127a + 128a | 2/1 | 52 |
| 5 | 37a | 126b | 1/2/10 | 7 | 127b + 128b | 1.4/1 | 83 |
| 6 | 37a | 126c | 1/2/10 | 7 | 127c + 128c | 2.7/1 | 70 |
| 7 | 37o | 126a | 1/2/10 | 7 | 127d + 128dd | Trace/1 | 52 |
| 8 | 124 | 126a | 1/2/10 | 7 | 127a + 128a | 1/1.2 | 61 |
| 9 | 125 | 126a | 1/2/10 | 7 | 127a + 128a | 4/1 | 64 |
| 10 | 125 | 126b | 1/2/10 | 7 | 127b + 128b | 2/1 | 59 |
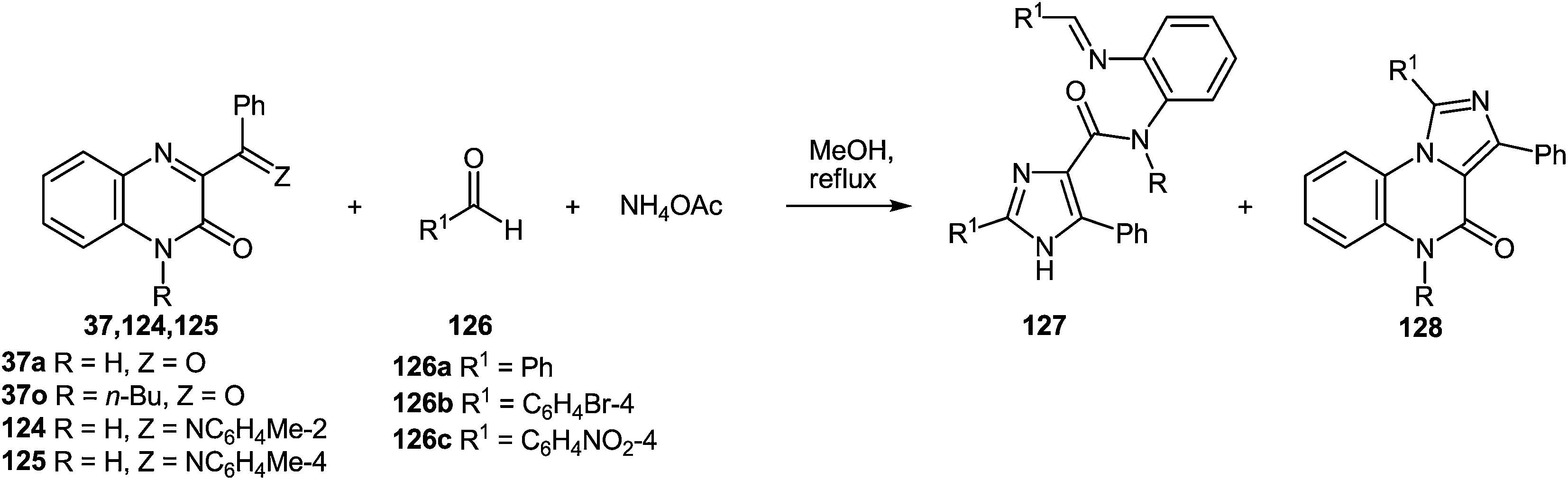
To determine the scope and generality of this reaction, various substituted aldehydes and 3-pyridinecarboxaldehyde were utilized. The desired products were obtained and the results are summarized in Table 10.
|
|
|||
|---|---|---|---|
| Entry | Substrate | R1 | Products (yield) |
| 1 | 126a | Ph | 127a + 128a |
| (49%) (26%) | |||
| 2 | 126b | C6H4Br-4 | 127b + 128b |
| (47%) (33%) | |||
| 3 | 126c | C6H4NO2-4 | 127c + 128c |
| (47%) (17%) | |||
| 4 | 126d | C6H4F-4 | 127e + 128e |
| (45%) (26%) | |||
| 5 | 126e | C6H4Cl-4 | 127f + 128f |
| (40%) (28%) | |||
| 6 | 126f | C6H4I-4 | 127g + 128g |
| (44%) (29%) | |||
| 7 | 126g | Py-3 | 127h + 128h |
| (38%) (42%) | |||
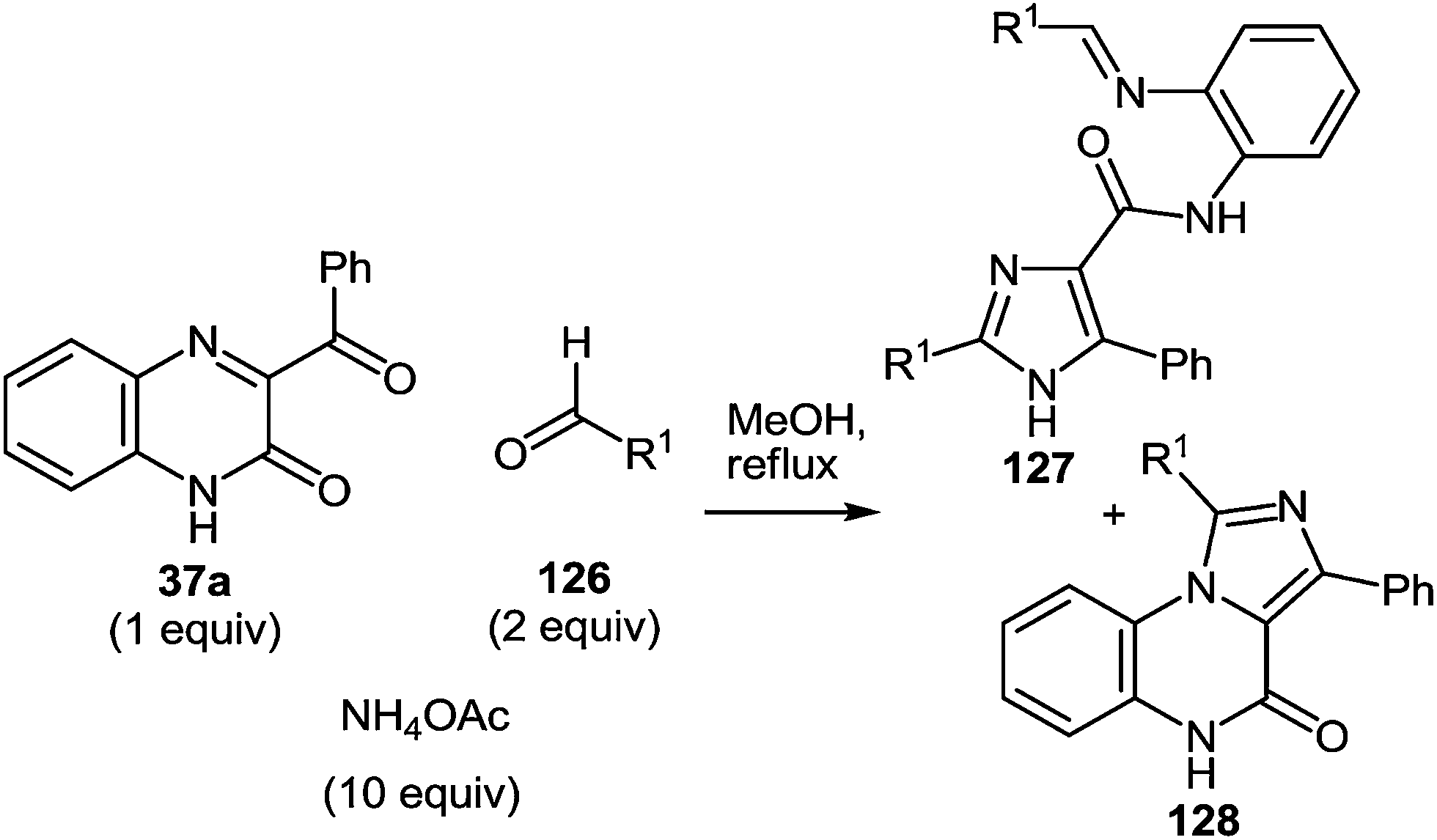
The presence of the o-iminoanilide substituent at position 4 of imidazoles 127 makes it possible to use them in further syntheses. Based on the four imidazole derivatives 127a, c, e, h it was shown that the reaction of these compounds with ammonium acetate in acetic acid proceeds with the formation of 2-(imidazol-4-yl)benzimidazoles 129a–d in almost quantitative yields (Table 11).
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | R1 | Product | Yield (%) |
| 1 | 127a | Ph | 129a | 94 |
| 2 | 127c | C6H4NO2-4 | 129b | 99 |
| 3 | 127e | C6H4F-4 | 129c | 98 |
| 4 | 127h | Py-3 | 129d | 95 |
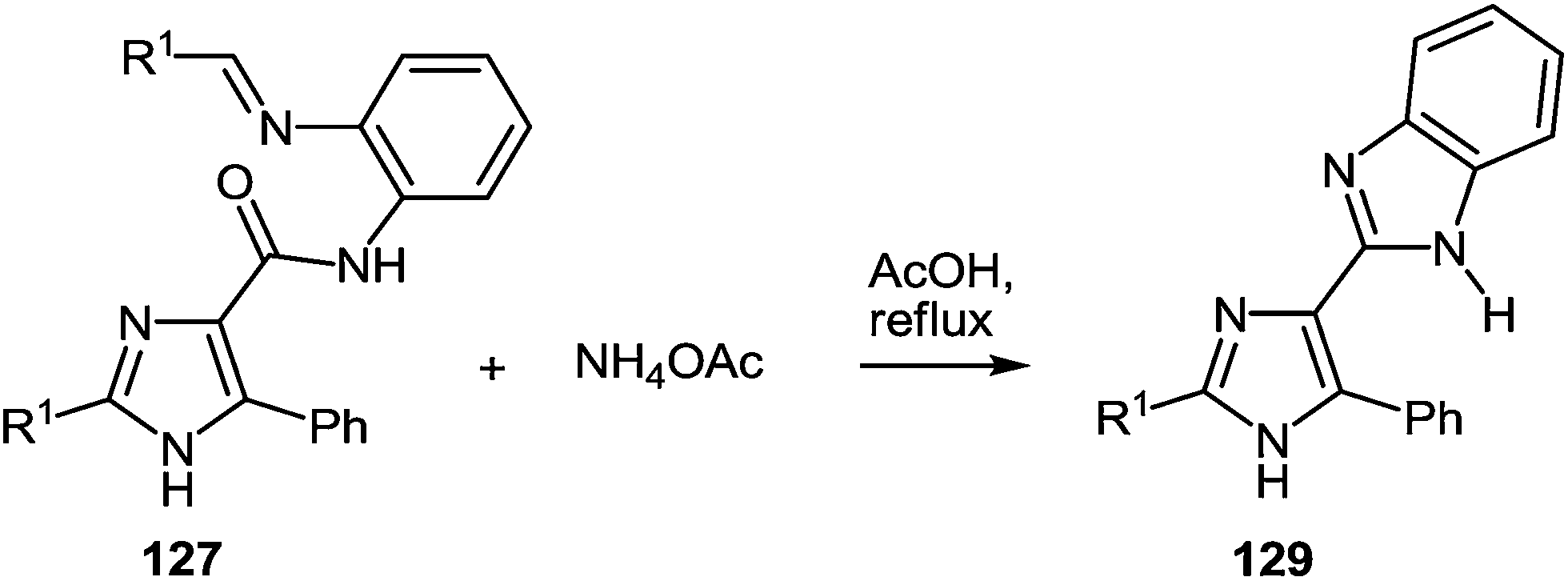
A plausible mechanism for the synthesis of imidazoles, imidazo[1,5-a]quinoxalin-4(5H)-ones and benzimidazoles has been proposed (Scheme 45). The formation of the diamine intermediate A takes place during the initial stage of the reaction. Intermediate A condenses with the 3-benzoylquinoxalin-2(1H)-one 37s followed by dehydration to afford the imino intermediate B, which is transformed in two different ways (pathway I and pathway II). Pathway I proceeds by cascade reactions involving: (a) acid-catalyzed ring-closure of intermediate C with the formation of spiro-compound D, (b) acid-catalyzed ring-opening of spiro-compound E with the formation of the imidazole derivative F, which is rearranged into the imidazole derivative Gvia a [1,5] hydrogen shift, and (c) reaction of the latter with the aldehyde to form compound 127. Pathway II involves the tautomerism of intermediate B with the formation of compound H, which under acid catalysis undergoes intramolecular cyclization to give I. The final product 128 is formed following the elimination of ammonia through intermediates J and K.
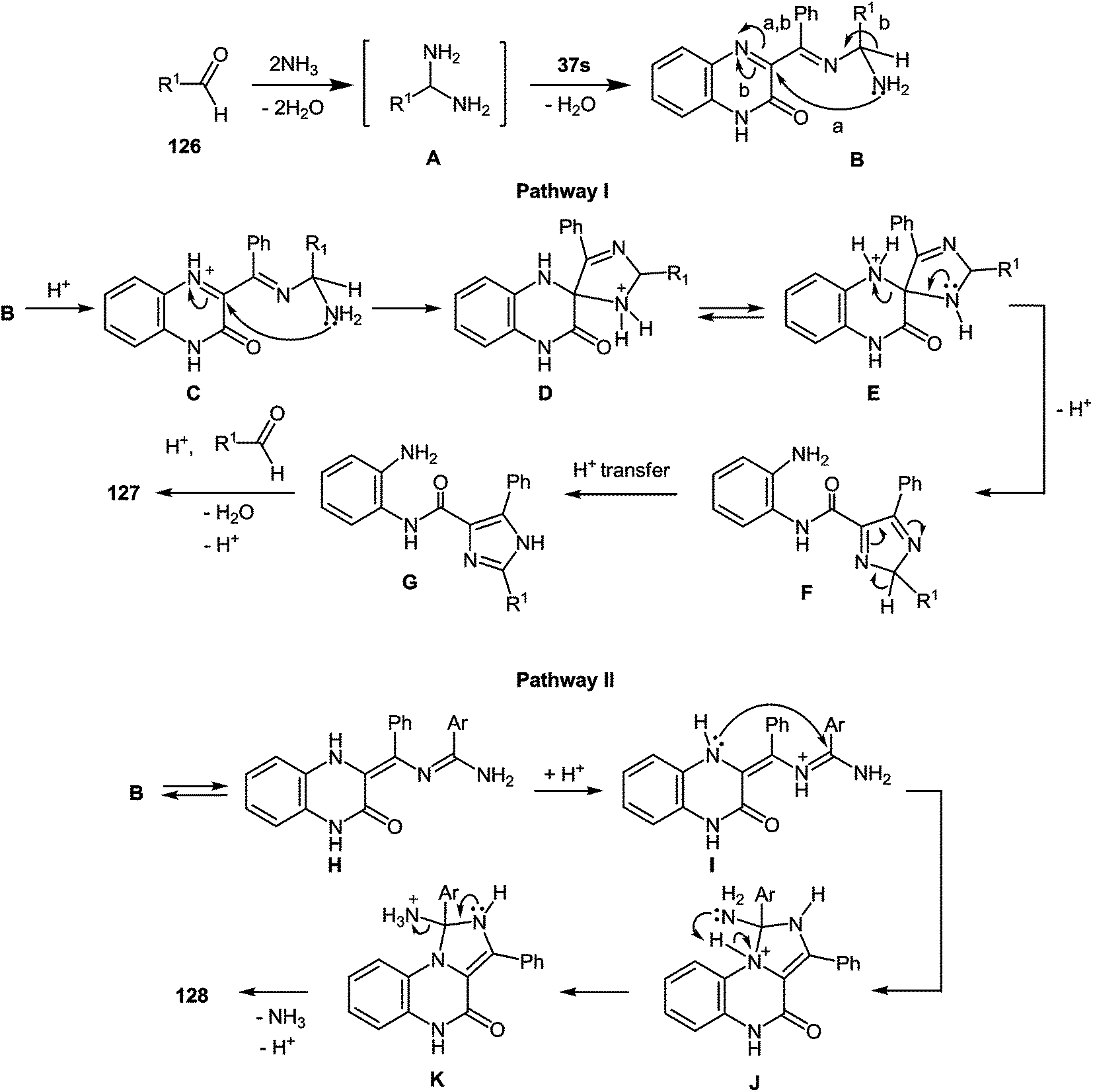 | ||
| Scheme 45 A plausible mechanism for the formation of imidazoles. Pathway I – acid catalysis through ring-closure and ring-opening processes. Pathway II – via a novel acid catalysis imidazoannulation of quinoxalin-2(1H)-ones. | ||
It is apparent that the formation of 2-(imidazol-4-yl)benzimidazoles 129a–d involves the ammonolysis of imidazoles 127a, c, d, g to form the corresponding o- aminoanilide derivative G as the first step. The next step involves an intramolecular nucleophilic attack by the amino group on the carbonyl group with the formation of the intermediate hydroxy-derivative L, and then the elimination of water. This leads to the formation of the final product 129 (Scheme 46).
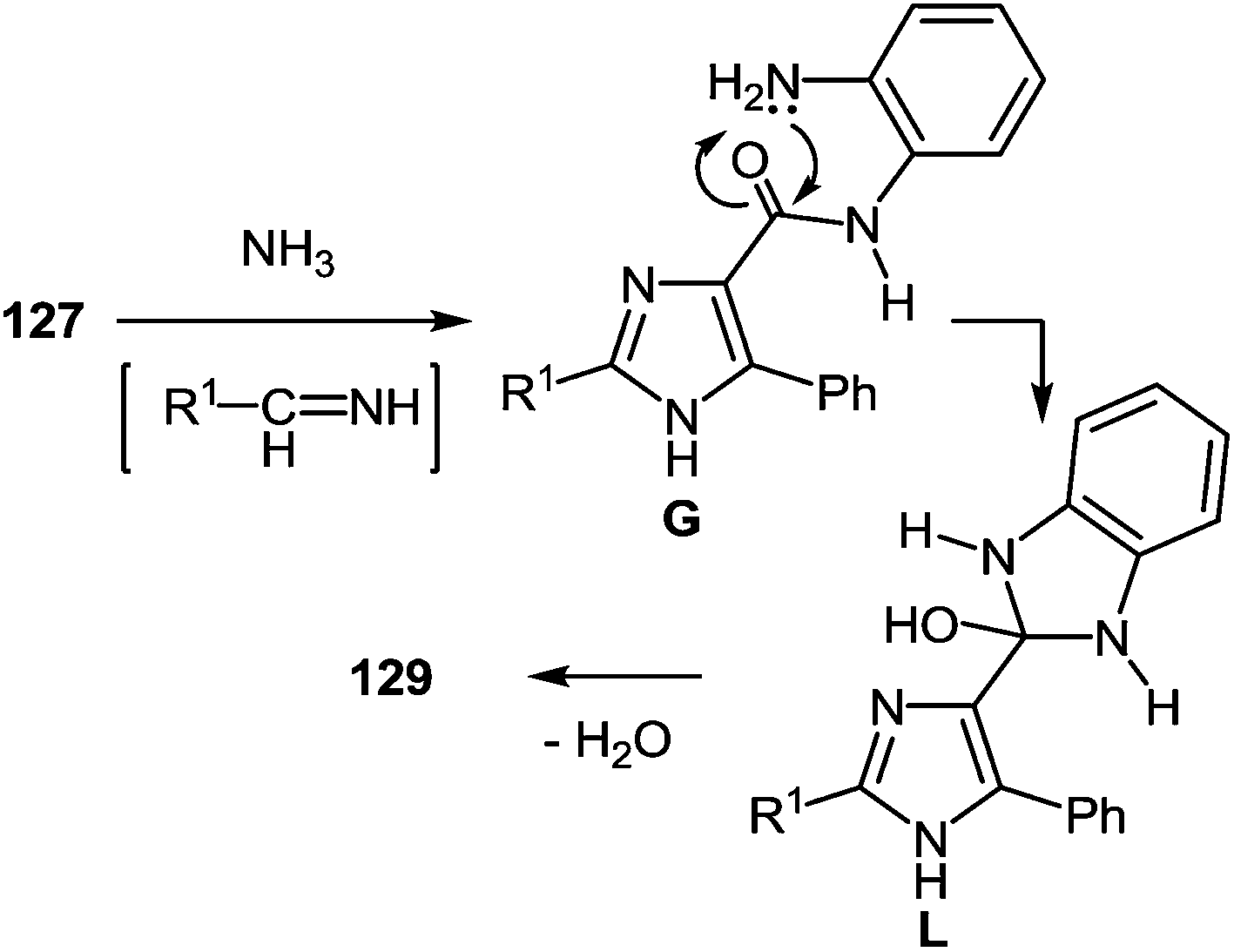 | ||
| Scheme 46 A possible mechanism for the formation of 2-(imidazol-4-yl)benzimidazoles 129. | ||
This was accomplished by the novel ring-opening of 3-aroylquinoxalin-2(1H)-ones on exposure to diaminoarylmethanes intermediately generated from arylaldehydes and ammonium acetate. The introduction of the o-iminoanilide fragment at position 4 of the imidazole derivatives with the help of this method makes it possible to synthesize 2-(imidazol-4-yl)benzimidazoles. The simplicity of this method, high yields, easy work-up and purification of compounds by crystallization are the key advantages.
5 Synthesis of 2-(3-arylindolizin-2-yl)benzimidazole
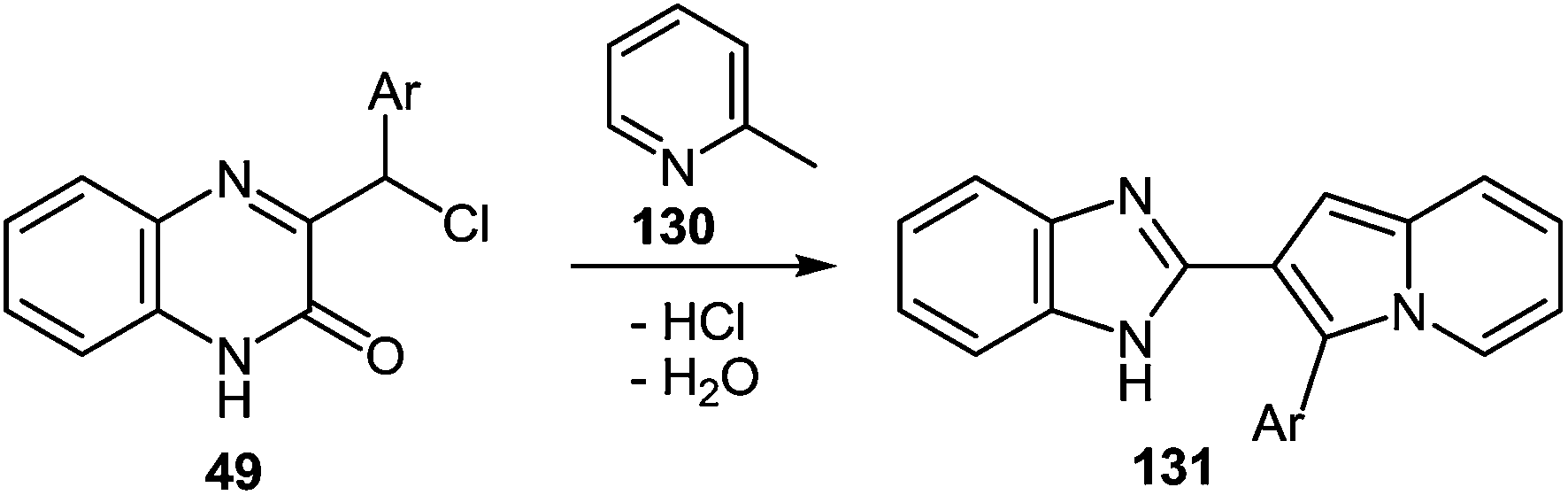
One of the common reactions of α-haloketones in the chemistry of heterocycles is the Chichibabin reaction, that is, the synthesis of indolizines by interaction of α-picoline with α-haloketones. It was carried out in a series of reactions according to Chichibabin. Thus, the reaction of quinoxalin-2-one 49 (see Subsection 2.3E) with α-picoline 130 at reflux results in high yields of the corresponding 2-(indolizinyl)benzimidazoles 131 (Table 12). As is evident from the structure of compounds 131, the C(2)–C(3)–C(Cl)Ar and NC–Me fragments of quinoxaline 49 and α-picoline 130 are involved in constructing the two new heterocyclic rings.89,107a,109
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Ar | Product | Yield (%) |
| 1 | 49a | C6H4NO2-4 | 131a | 72 |
| 2 | 49b | Ph | 131b | 78 |
| 3 | 49c | C6H3-di-Cl-2,4 | 131c | 76 |
| 4 | 49d | C6H3-di-Cl-3,5 | 131d | 65 |
| 5 | 49e | CH2Ph | 131e | 70 |
Initially, a complete dissolution of compounds 49 is observed in refluxing the α-picoline solution. After that there rapidly occurs an abundant precipitation of crystals which gradually dissolve during the course of the reaction. The yield of crystalline products with a precise melting point, are obtained, for example, after refluxing quinoxalin-2(1H)-one 49c in α-picoline 130 for 1 h, and the yield is 41% (Table 13).
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Ar | Product | Yield (%) |
| 1 | 49a | C6H4NO2-4 | 132a | 10 |
| 2 | 49b | Ph | 132b | 37 |
| 3 | 49c | C6H3-di-Cl-2,4 | 132c | 41 |
| 4 | 49d | C6H3-di-Cl-3,5 | 132d | 16 |
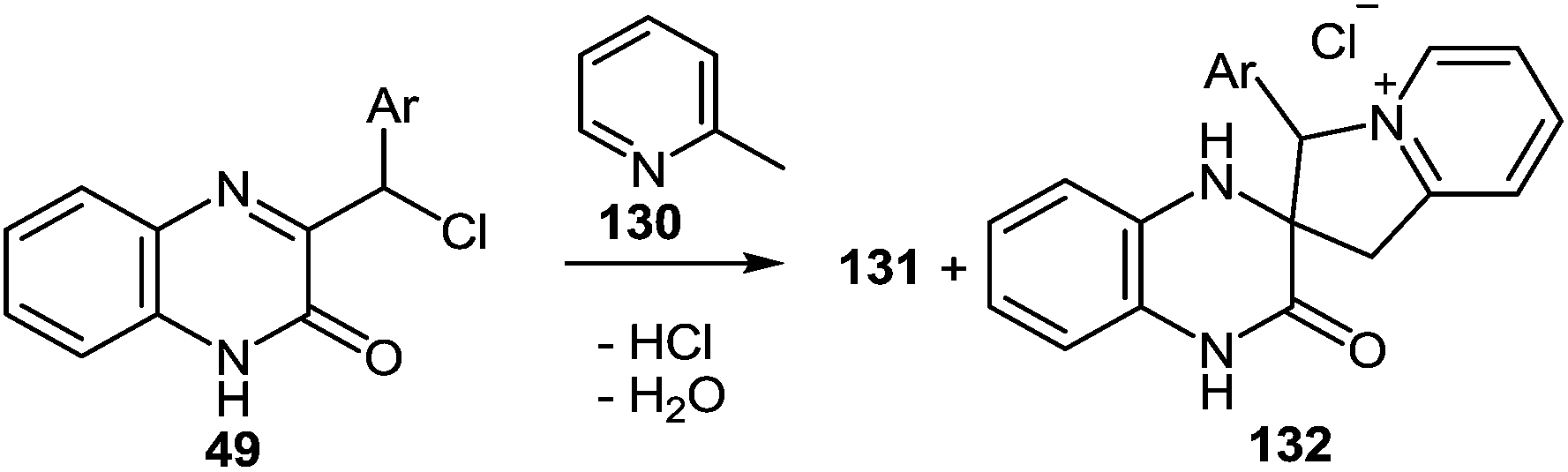
It should be noted that spiro-compound 132 is quantitatively transformed into 2-(3-arylindolizin-2-yl)benzimidazole 131 not only in boiling α-picoline, but also in acetic acid (Scheme 47).
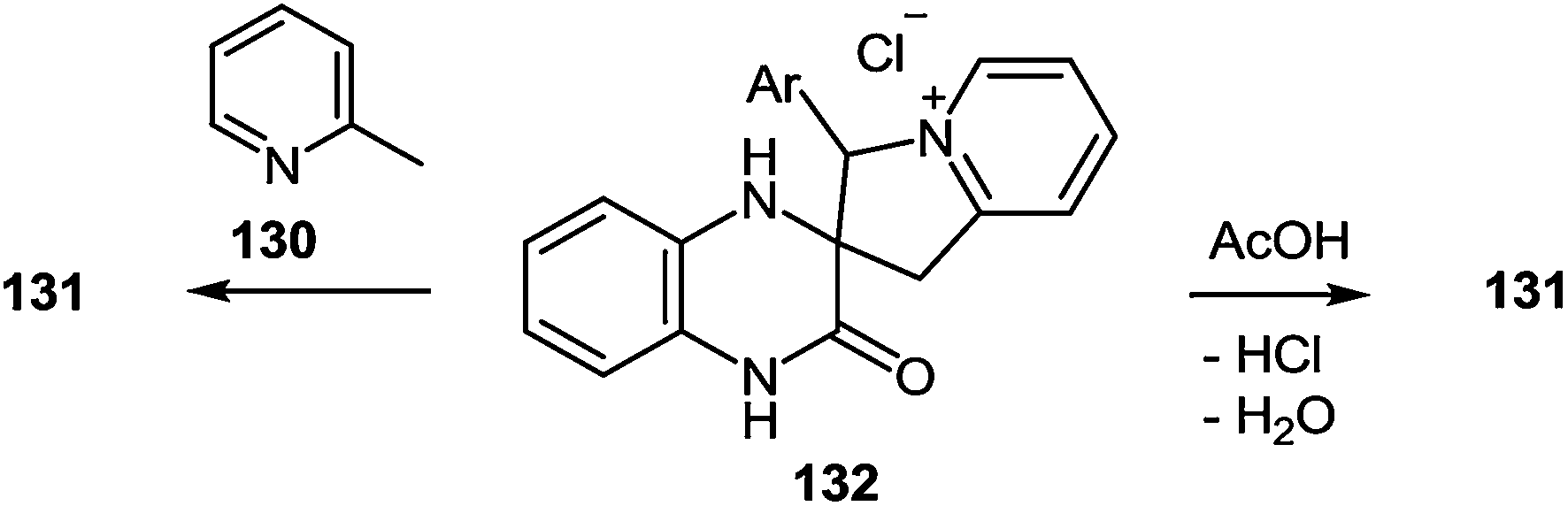 | ||
| Scheme 47 The rearrangement of spiro-quinoxalinones 132. | ||
A first-stage product resulting from the nucleophilic displacement of the Cl atom by the pyridine N atom of α-picoline under milder reaction conditions than are required for the formation of a spiro-compound or its rearrangement into a benzimidazole derivative was isolated and characterized. For instance, when 3-(α-chlorobenzyl)quinoxalin-2(1H)-one 49a was stirred in α-picoline 130 at 50 °C for 3 h (Scheme 48), it was possible to obtain the first stage product of the reaction, that is, compound 133, which in boiling acetic acid is transformed into a rearrangement product 132 in quantitative yields.89
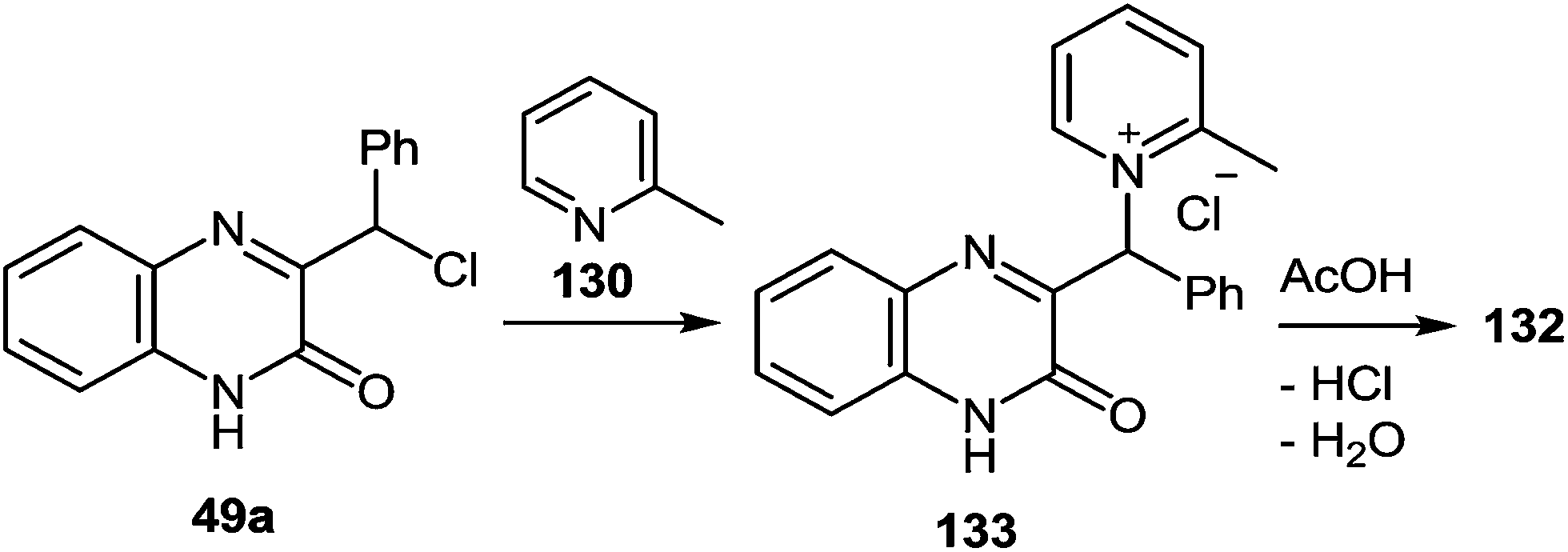 | ||
| Scheme 48 The synthesis and rearrangement of 2-methyl-1-[(3-oxo-3,4-dihydroquinoxalin-2-yl)(phenyl)methyl]pyridinium chloride 133. | ||
The formation of the rearrangement product can be represented by Scheme 49. According to this scheme, the initial step involves the nucleophilic displacement of the Cl atom by the pyridine N atom, which is followed by a cascade transformation. The latter involves: (a) dehydrochlorination with the abstraction of a methyl H atom of α-picoline, (b) intramolecular nucleophilic addition of the methylidene C atom of the α-picoline fragment to the azomethine C atom of the quinoxaline system (spiro-compound B), (c) cleavage of the C(3)–N(4) bond (N-substituted 1,2-DAB C), (d) closure of a five-membered ring (benzimidazoline D), and (e) elimination of water.
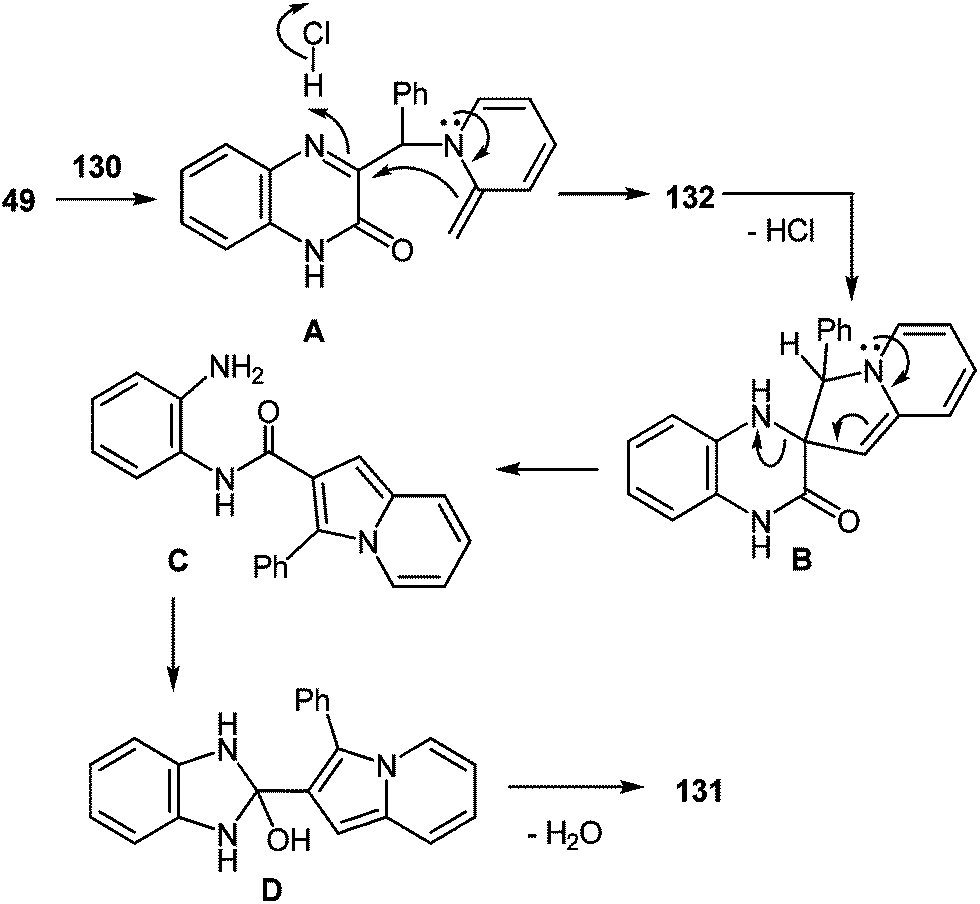 | ||
| Scheme 49 Proposed mechanism of the rearrangement. | ||
According to this scheme, the initial step involves the nucleophilic displacement of the Cl atom by the pyridine N atom, which is followed by a cascade transformation. The latter involves: (a) dehydrochlorination with the abstraction of a methyl H atom of α-picoline, (b) intramolecular nucleophilic addition of the methylidene C atom of the α-picoline fragment to the azomethine C atom of the quinoxaline system (spiro compound B), (c) cleavage of the C(3)–N(4) bond (N-substituted 1,2-DAB C), (d) closure of a five-membered ring (benzimidazoline D), and (e) elimination of water.
3-(α-Haloarylmethyl)- and 3-(α-halophenethyl)quinoxalin-2(1H)-ones, as well as their various derivatives have been shown to react with α-picolines, providing high yields of 3-aryl- and 3-alkylindolizinylbenzimidazoles.
6 Synthesis of 2-(pyrazol-3-yl)benzimidazoles
One of the most popular reactions of β-diketones in heterocyclic chemistry is the Knorr reaction, that is, the synthesis of pyrazoles by interaction of β-diketones with hydrazines. It has been described that the interaction of 3-(arylacylidene)-3,4-dihydroquinoxalin-2(1H)-ones 50 (see Subsection 2.3E) with hydrazine hydrate in boiling butanol solution proceeds with the formation of 3′-aryl-1,2,3,4,4′,5′-hexahydro[quinoxalin-2,5′-pyrazol]-3-ones 134a–f in good yields (Table 14).76a,107a,110|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | Ar | Product | Yield (%) |
| 1 | 50a | H | H | Ph | 134a | 81 |
| 2 | 50b | Me | Me | Ph | 134b | 76 |
| 3 | 50c | H | Cl | Ph | 134c | 64 |
| 4 | 50d | NO2 | H | Ph | 134d | 73 |
| 5 | 50e | H | H | C6H4Me-4 | 134e | 77 |
| 6 | 50f | H | H | C6H4Cl-4 | 134f | 73 |
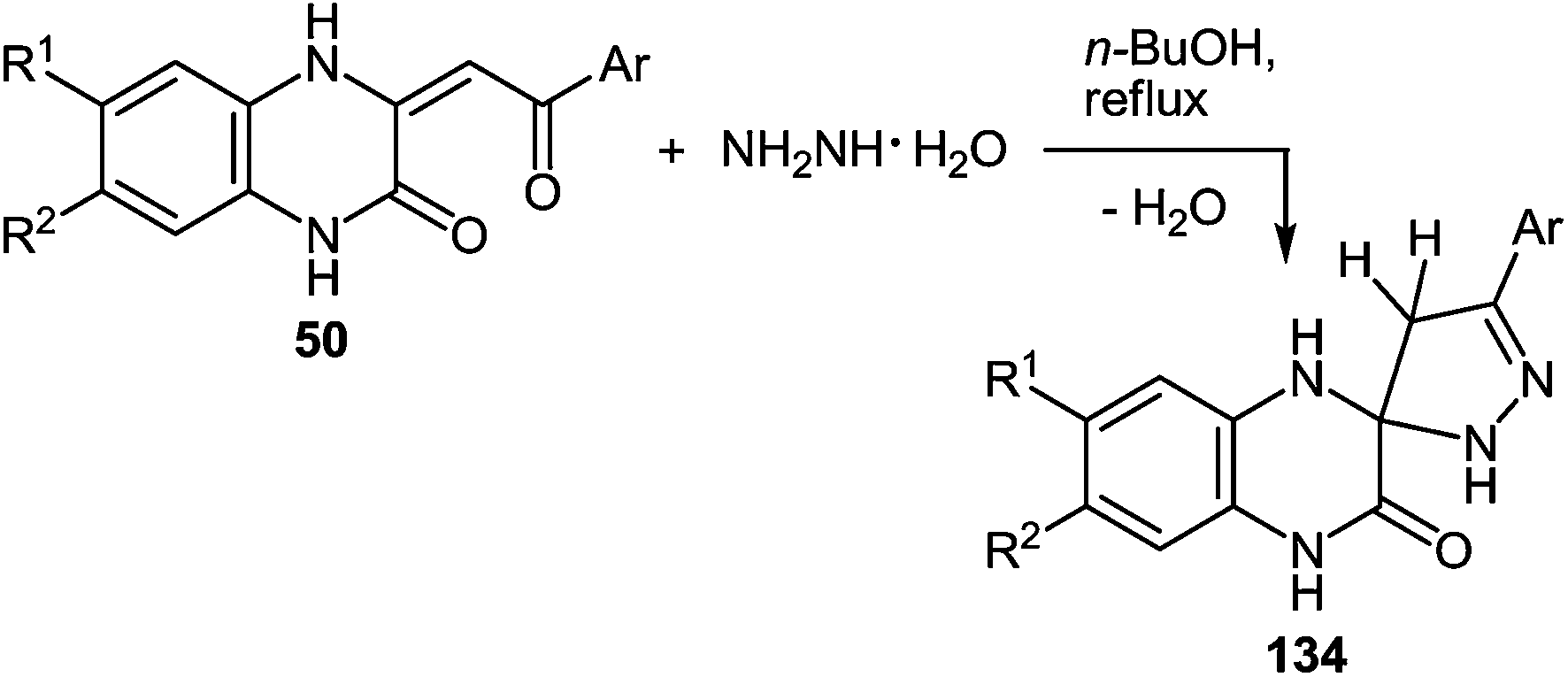
Boiling spiro-quinoxalin-2,5′-pyrazol-3-ones 134a–f in an acetic acid solution for 8 h results in the acid catalysis rearrangement with the release of water and the formation of corresponding 2-(pyrazol-3′-yl)benzimidazoles in quantitative yields (Table 15).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | Ar | Product | Yield (%) |
| 1 | 134a | H | H | Ph | 135a | 99 |
| 2 | 134b | Me | Me | Ph | 135b | 99 |
| 3 | 134c | H | Cl | Ph | 135c | 98 |
| 4 | 134d | NO2 | H | Ph | 135d | 96 |
| 5 | 134e | H | H | C6H4Me-4 | 135e | 99 |
| 6 | 134f | H | H | C6H4Cl-4 | 135f | 85 |
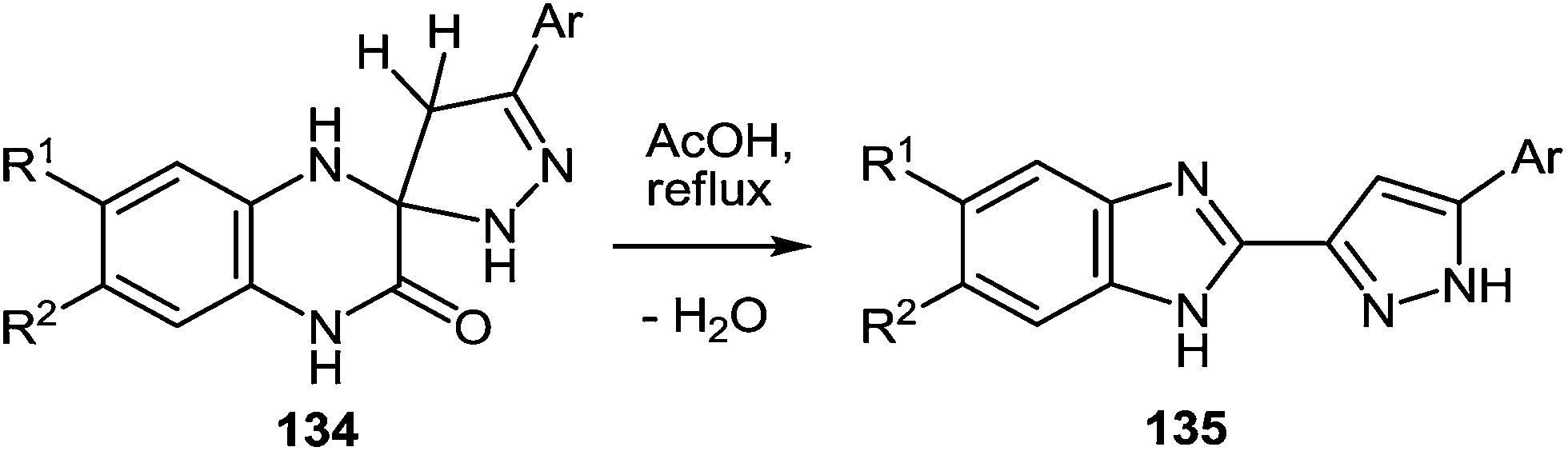
Thus, both the formation of spiro-quinoxalinones 134a–f and their rearrangement into the corresponding pyrazolylbenzimidazoles 135a–f proceed in high yields (Table 14 and 15). As is evident from the structure of compounds 135a–f, the C(2)–C(3)CH–C(O)–Ar fragment of quinoxalines 50a–f and hydrazine hydrate are involved in the formation of the heterocyclic systems.
On the basis of the known chemistry of hydrazines,111 ketones,112 and quinoxalinones105 it is reasonable to assume that the first stage of the reaction mechanism involves the addition of the hydrazine to the carbonyl group of the 3-arylacylidene fragment of quinoxalin-2(1H)-one 50 with the formation of intermediate A capable of reversible tautomerization to intermediate B. The next step involves a nucleophilic attack of the amino group on the C(3) atom of the quinoxalin-2(1H)-one to form the spiro-quinoxaline derivative 134. Rearrangement of the spiro-quinoxaline is then assumed to occur according to Scheme 50, which proceeds by cascade reactions involving: (a) acid-catalyzed ring-opening in spiro-compound C with the intermediate formation of pyrazolo derivative D, (b) intramolecular nucleophilic attack by the amino group on the carbamoyl carbonyl group with the intermediate formation of hydroxy-derivative E, and (c) elimination of water leading to the formation of the final product 135. It was shown that the reaction does not proceed in neutral or aprotic solvents.
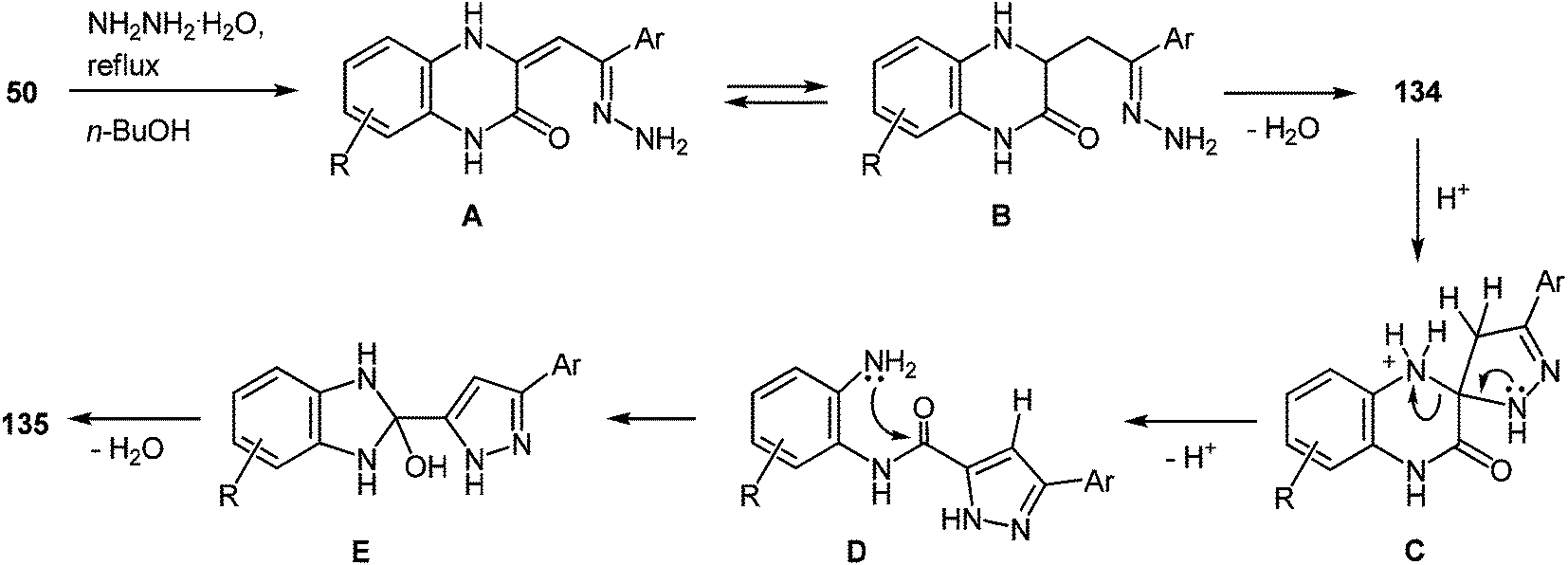 | ||
| Scheme 50 Proposed mechanism of the rearrangement with the initial attack on the aroyl carbonyl group. | ||
It should be pointed out that the formation of spiro-quinoxaline derivative 134 could be due to the Michael addition of hydrazine to the partially positive C(3) atom of the quinoxalin-2(1H)-one 50 in the first stage of the reaction mechanism with the formation of intermediate A′ capable of reversible tautomerization to intermediate B′. Then cyclization involves the nucleophilic attack of the amino group on the carbonyl group of the 3-arylacylidene fragment of quinoxalin-2(1H)-one (Scheme 51).
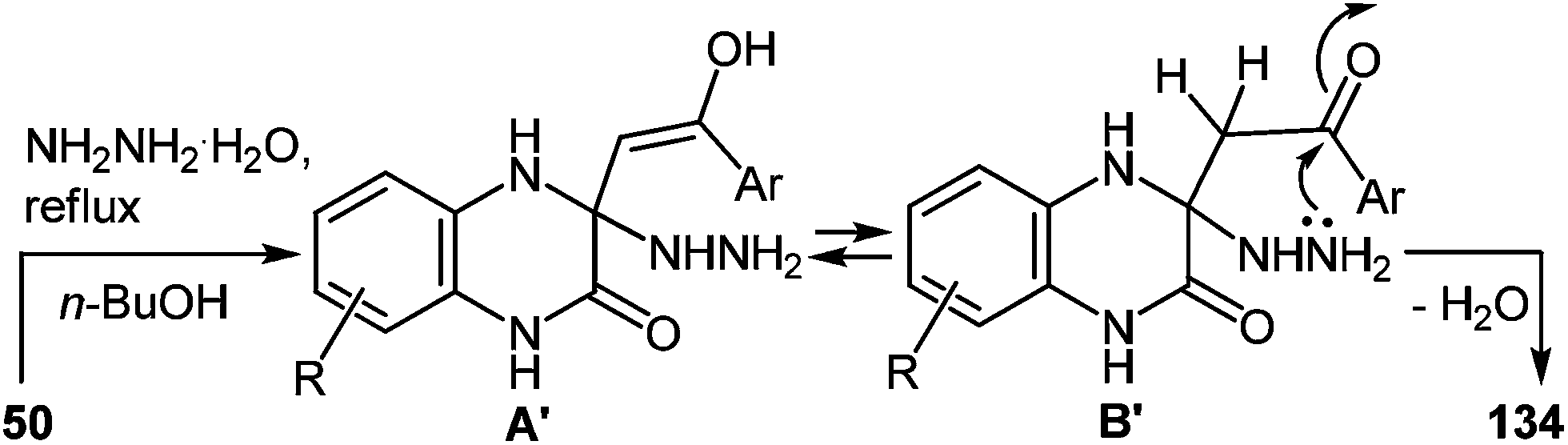 | ||
| Scheme 51 Proposed mechanism of the rearrangement with the initial attack on the C(3) atom of the quinoxalin-2(1H)-one. | ||
It is worth noting that the reaction of phenylhydrazine with 3-arylacylidene-3,4-dihydroquinoxalin-2(1H)-one 50a proceeds similarly to the reaction with hydrazine hydrate. This involves the formation of spiro-compound 136, which rearranges into pyrazolylbenzimidazole 137, and no other possible regioisomer 137a, in boiling acetic acid (Scheme 52).
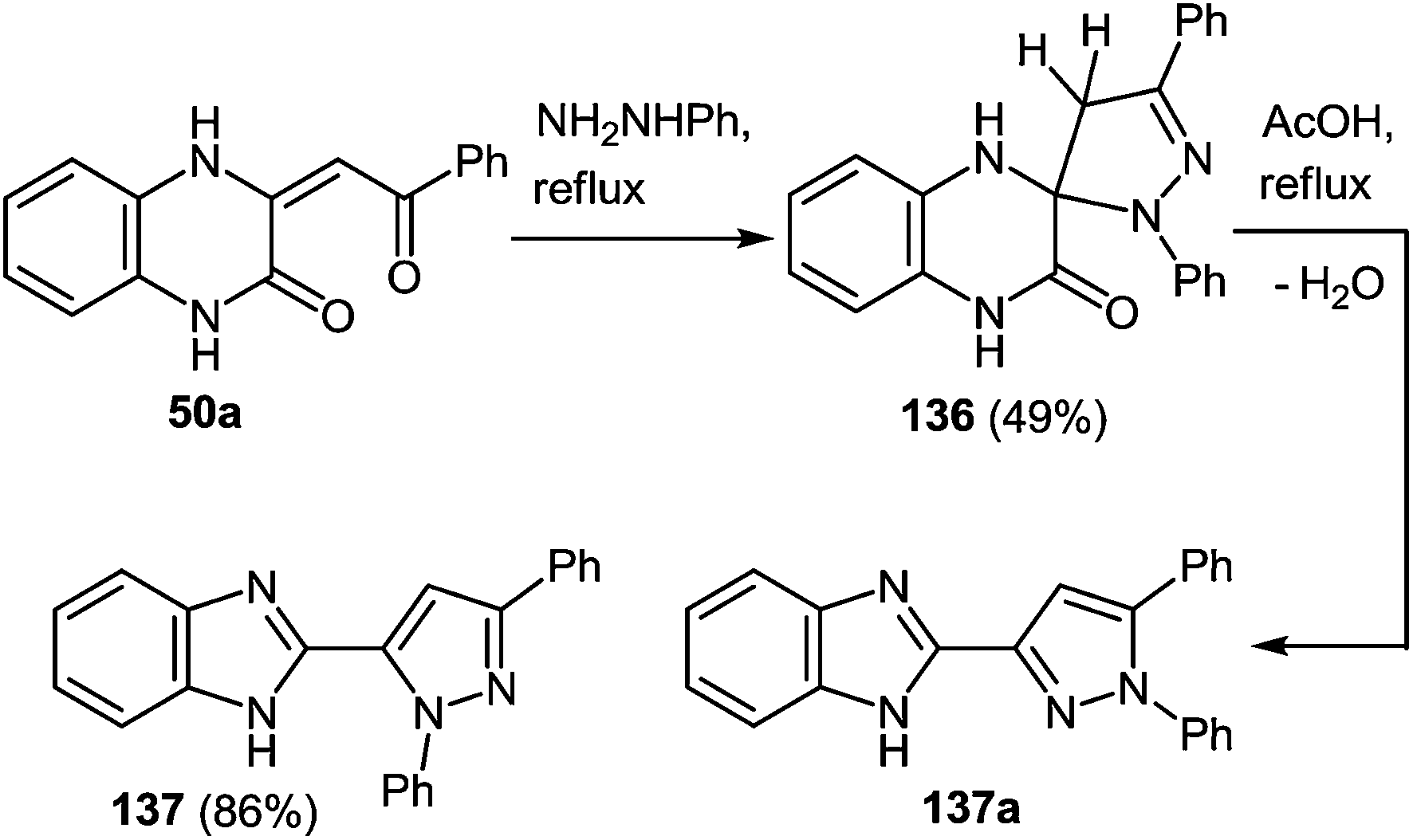 | ||
| Scheme 52 Phenylhydrazine as nucleophilic reagent in the rearrangement of 3-arylacylidene-3,4-dihydroquinoxalin-2(1H)-one. | ||
This was accomplished via a novel quinoxalinone-benzimidazole rearrangement of 3-arylacylidene-3,4-dihydroquinoxalin-2(1H)-ones on exposure to hydrazine hydrate and phenylhydrazine. The reaction is readily applicable to large scale synthesis.
7 Synthesis of 2-(pyrrol-3-yl)benzimidazole
One of the most popular reactions of α-aminoketones in heterocyclic chemistry is another Knorr reaction, that is, the synthesis of pyrroles by the interaction of α-aminoketones with β-dicarbonyl compounds. Assuming that 3-(α-aminobenzyl)quinoxalin-2-(1H)one is the very analog of α-amineketones we have carried out the reaction of 3-(α-aminobenzyl)quinoxalin-2-(1H)-one hydrochloride 51 (see Subsection 2.3E) with acetoacetic ester 138 in the boiling acetic acid. Reaction proceeds with the contraction of the pyrazine ring system as a result of the rearrangement involving C(2)-C(3)-C(NH2)Ph and the C(2)-C(3) fragments of the quinoxalinone system and acetoacetic ester, respectively, with the formation of 2-(5-methyl-2-phenyl-4-ethoxycarbonyl-pyrrol-3-yl)benzimidazole 139 (Scheme 53).76b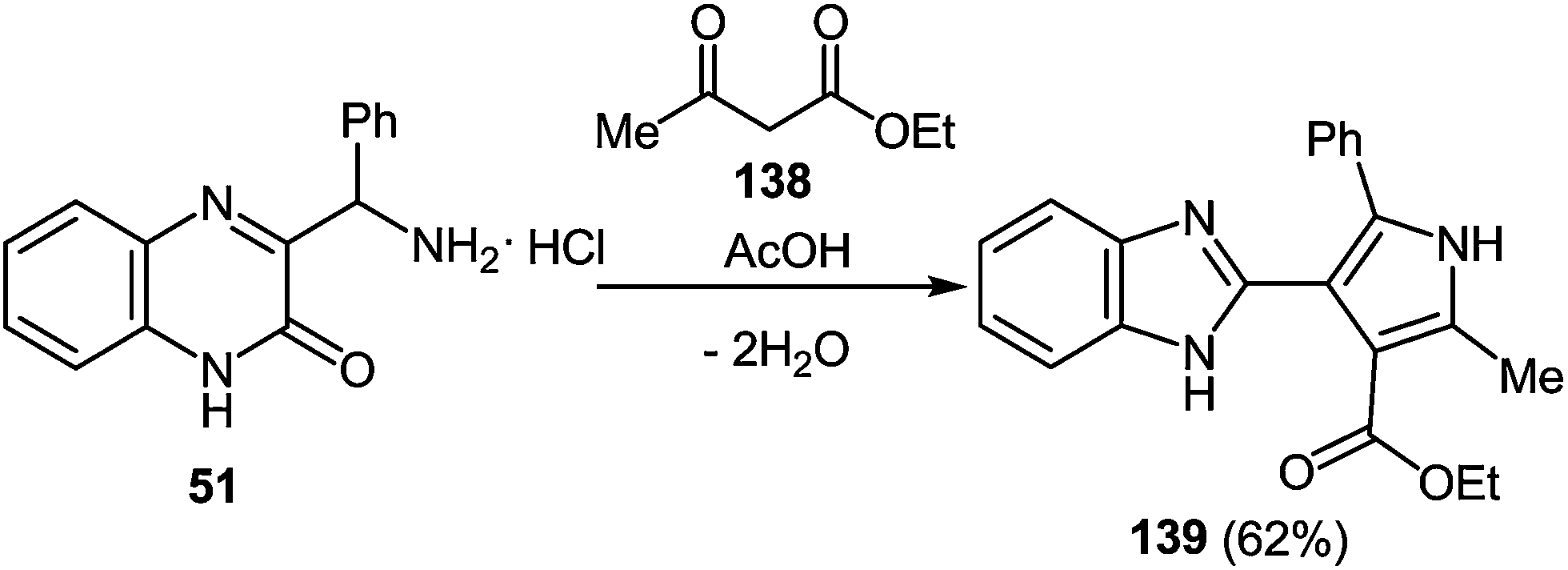 | ||
| Scheme 53 The rearrangement of 3-(α-aminobenzyl)quinoxalin-2-(1H)-one hydrochloride in the reaction with acetoacetic ester. | ||
Instead of the 3-(α-aminobenzyl)quinoxalin-2-(1H)-one 51 hydrochloride in the reaction with acetoacetic ester 138 its free base is used. This has no significant effect on the yield of the product of the rearrangement, which is apparently associated with the occurrence of various acid catalyzed side reactions involving acetoacetic ether.
To improve the yield of the product of the rearrangement the main principle of this rearrangement, proposed above was used (see Subsection 2.3D). In accordance with the above principle the spiro[4-pyrrolyne-3,2′-quinoxalin]-3′(4′H)-one 140 was synthesized. The 3-(α-aminobenzyl)quinoxalin-2(1H)-one 51, was considered to be the hetero analogue of α-aminocarbonyl compound according to the Knorr reaction113 (i.e. obtaining pyrroles by the condensation of α-aminoketones with ketones containing an activated methylene group). Thus, at position 2 of quinoxalinone 51 the necessary pyrrolidine system has been set up with the help of the reaction of the compound 51 with acetoacetic ester 138 in EtOH in the presence of KOH. The reaction proceeds smoothly with the formation of the desired spiro-compound – 5-methyl-2-phenyl-4-ethoxycarbonyl-1′H-spiro[4-pyrrolyne-3,2′-quinoxalin]-3′(4′H)-one 140 with high yields (88%) (Scheme 54). It should be noted that the reaction proceeds well enough both at room temperature (12 h) and by reflux (4 h).
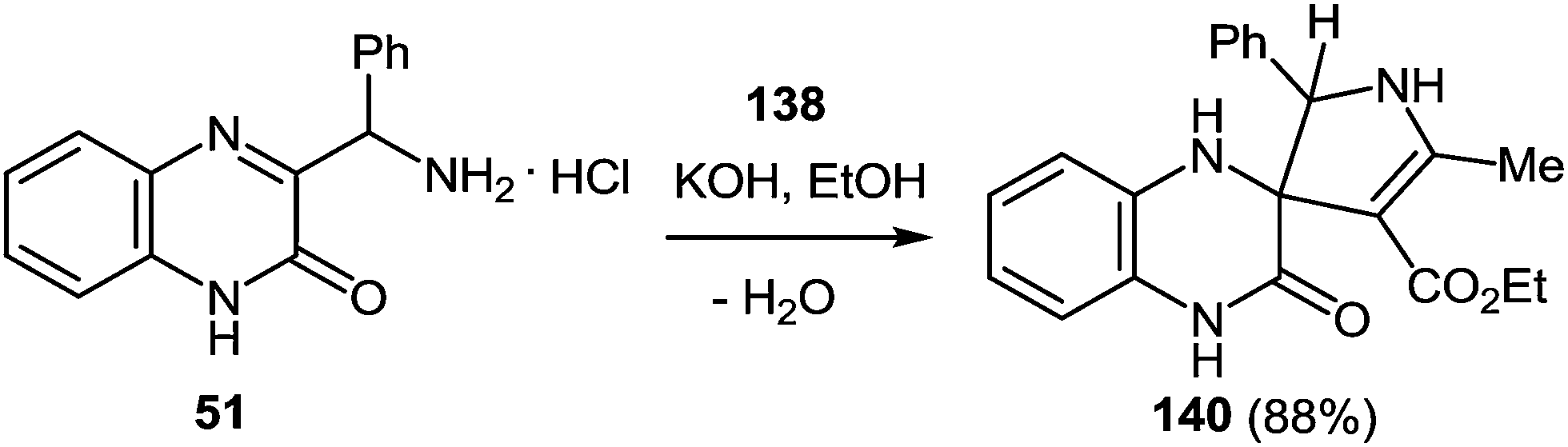 | ||
| Scheme 54 The synthesis of spiro[4-pyrrolyne-3,2′-quinoxalin]-3′(4′H)-one. | ||
Boiling the compound 140 in acetic acid for 1 h leads to the expected benzimidazole 139 in quantitative yields (Scheme 55).
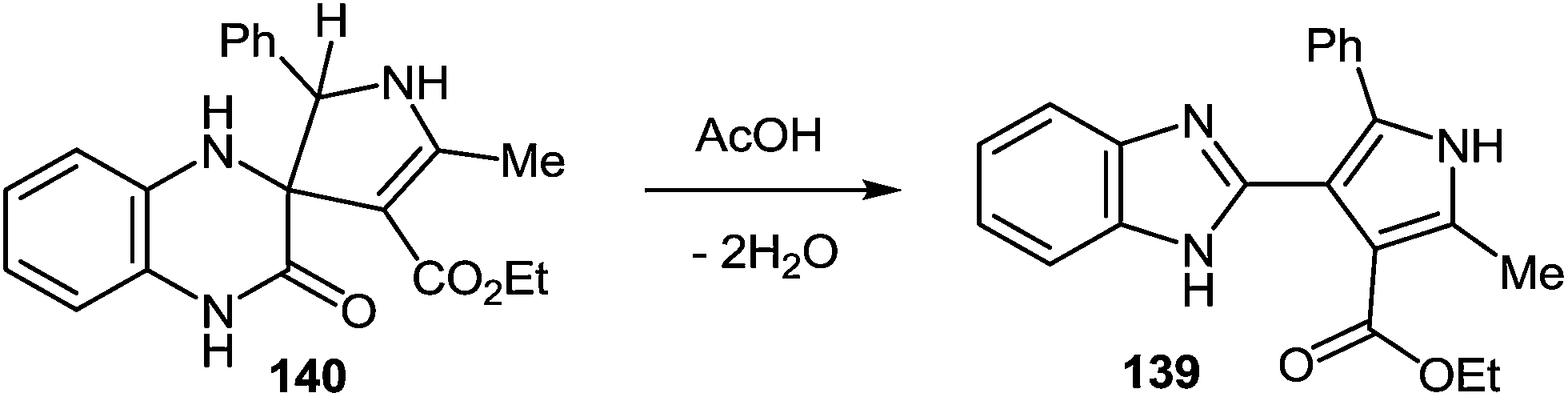 | ||
| Scheme 55 Acid-catalyzed rearrangement of spiro[4-pyrrolyne-3,2′-quinoxalin]-3′(4′H)-one 140. | ||
The formation of the rearrangement product in the reaction of 3-(α-aminobenzyl)quinoxalin-2(1H)-one 51 with acetoacetic ester can be represented as shown below in Scheme 58. According to the scheme, the formation of spiro-compound 140 occurs at the initial stage. In reaction conditions it further undergoes acid-catalyzed rearrangement. The latter involves the disclosure of the pyrazine ring of the quinoxalin-2(1H)-one system on the N(1)–C(2) bond in the intermediate salt A and the closure of the imidazole ring of the benzimidazole system with the newly formed amino group and the carbonyl group of carbamoyl fragment in the pyrrole derivative B (Scheme 56).
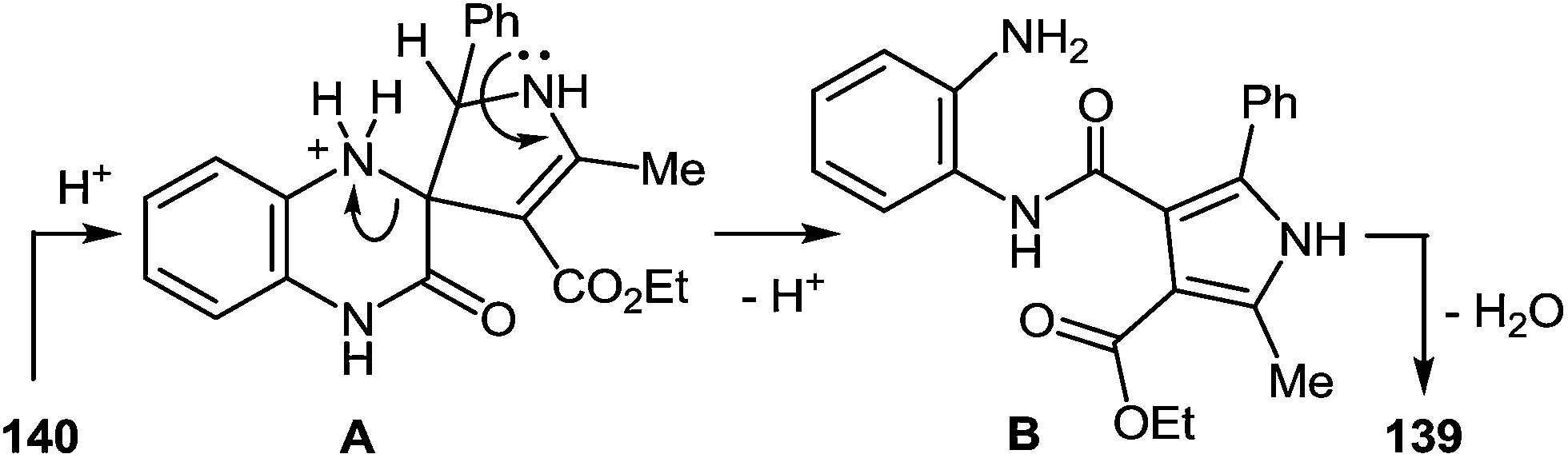 | ||
| Scheme 56 Proposed mechanism of the rearrangement. | ||
It should be pointed out that other substituted derivatives of 3-(α-aminobenzyl)quinoxalin-2(1H)-one also successfully react not only with acetoacetic ester but with other esters of β-oxoacids, resulting in corresponding derivatives of 2-(pyrrol-3-yl)benzimidazole in high yields.
8 Synthesis of 2-(benzimidazol-2-yl)quinolines
One of the most popular reactions of usual ketones in the heterocyclic chemistry is the Friedländer reaction, that is, the synthesis of quinolines by their interaction with o-acylarylamines in basic or acid catalysis conditions.This reaction has been modified according to the system based on aromatic aldehydes 126 and 3-methylquinoxalinones 52 (see Subsection 2.3E), easily available from methylpyruvates and 1,2-DABs. The first stage of the modification involves the formation of o-nitrostyryl derivatives of quinoxalin-2(1H)-one 141 (Scheme 57).107c
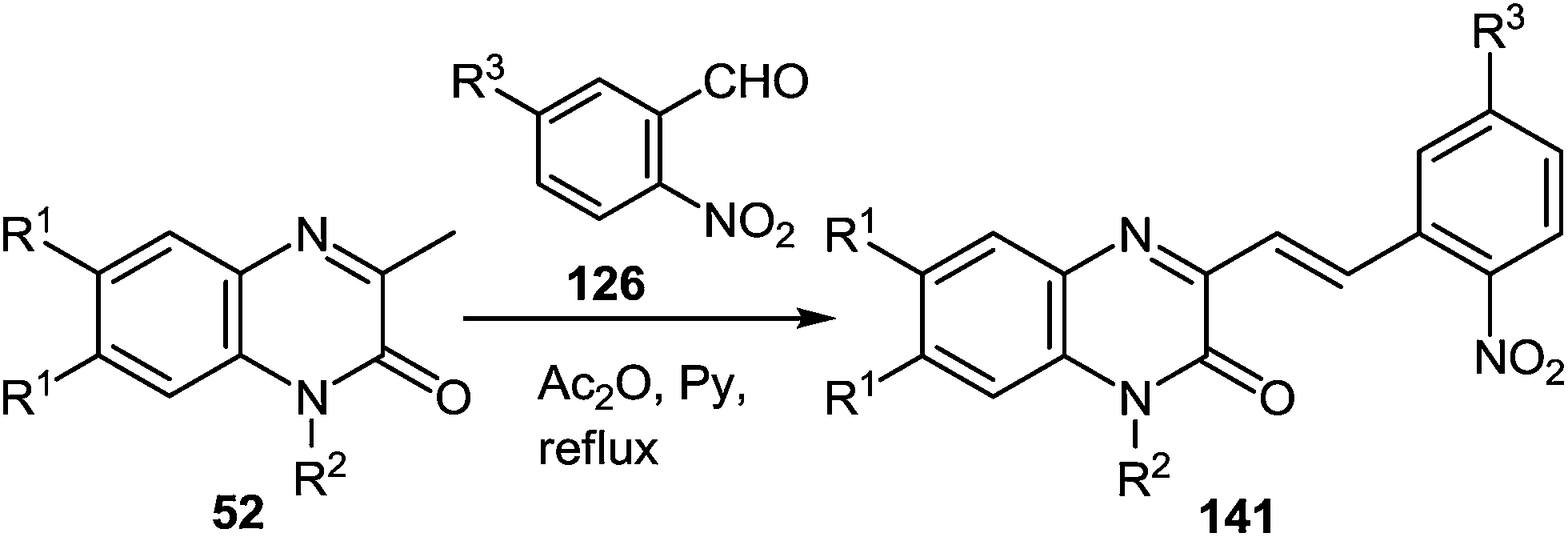 | ||
| Scheme 57 Synthesis of 3-(β-2-nitrostyryl)quinoxalin-2(1H)-ones. | ||
The second stage is based on the reduction of o-nitrostyryl derivatives 141 when exposed to Na2S2O4 (sodium hydrosulfate, sodium dithionate). O-Aminonitrostyryl derivatives of quinoxalin-2(1H)-one, obtained in these conditions immediately undergo acid-catalyzed rearrangement with the formation of corresponding benzimidazolyl quinoline derivatives. As is evident from the structure of compounds 142, the C(2)–C(3) fragment and the β-2-nitrostyryl substitute at position 3 of the quinoxalin-2(1H)-one system, are involved in constructing two new heterocyclic systems (Table 16).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | R3 | Product | Yield (%) |
| 1 | 141a | H | H | H | 142a | 80 |
| 2 | 141b | H | Me | H | 142b | 76 |
| 3 | 141c | H | Et | H | 142c | 74 |
| 4 | 141d | H | n-Pr | H | 142d | 53 |
| 5 | 141e | H | n-Bu | H | 142e | 69 |
| 6 | 141f | H | n-Pent | H | 142f | 70 |
| 7 | 141g | H | n-Hex | H | 142g | 54 |
| 8 | 141h | Me | H | H | 142h | 79 |
| 9 | 141i | H | H | Cl | 142i | 59 |
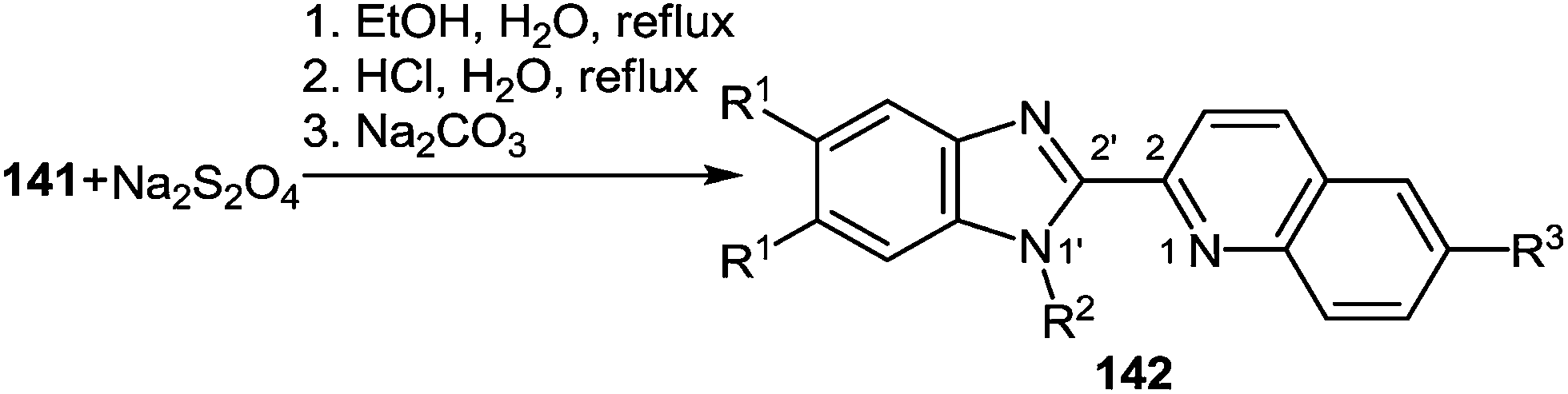
This reaction also proceeded with a compound 141j possessing two 3-(β-2-nitrostyryl)quinoxalin-2(1H)-one fragments, with the formation of a benzimidazole-monopodand with terminal benzimidazole fragments at C(2) and C(2′) positions of the quinoline ring system (Scheme 58).
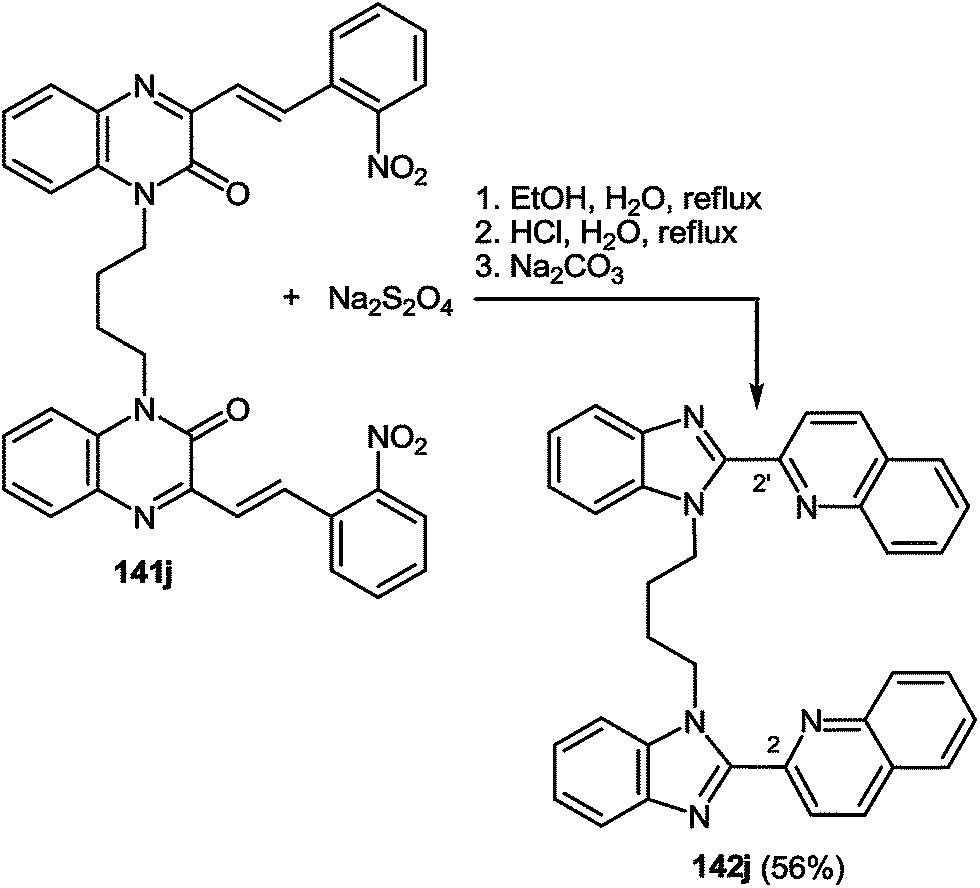 | ||
| Scheme 58 Synthesis of 1,4-bis[2-(quinolin-2-yl)-1H-benzimidazol-1-yl]butane. | ||
To investigate the reaction mechanism, the reduction of 3-(β-2-nitrostyryl)-6,7-dimethylquinoxalin-2(1H)-one 141h with hydrogen using a 10 mol% Pd/CaCO3 as the catalyst in methanol has been performed and the corresponding 3-(β-2-aminostyryl)-6,7-dimethylquinoxalin-2(1H)-one 143h obtained. When boiled in AcOH for 3 h the latter was transformed into 2-(benzimidazol-2-yl)quinoline 142h (Scheme 59).
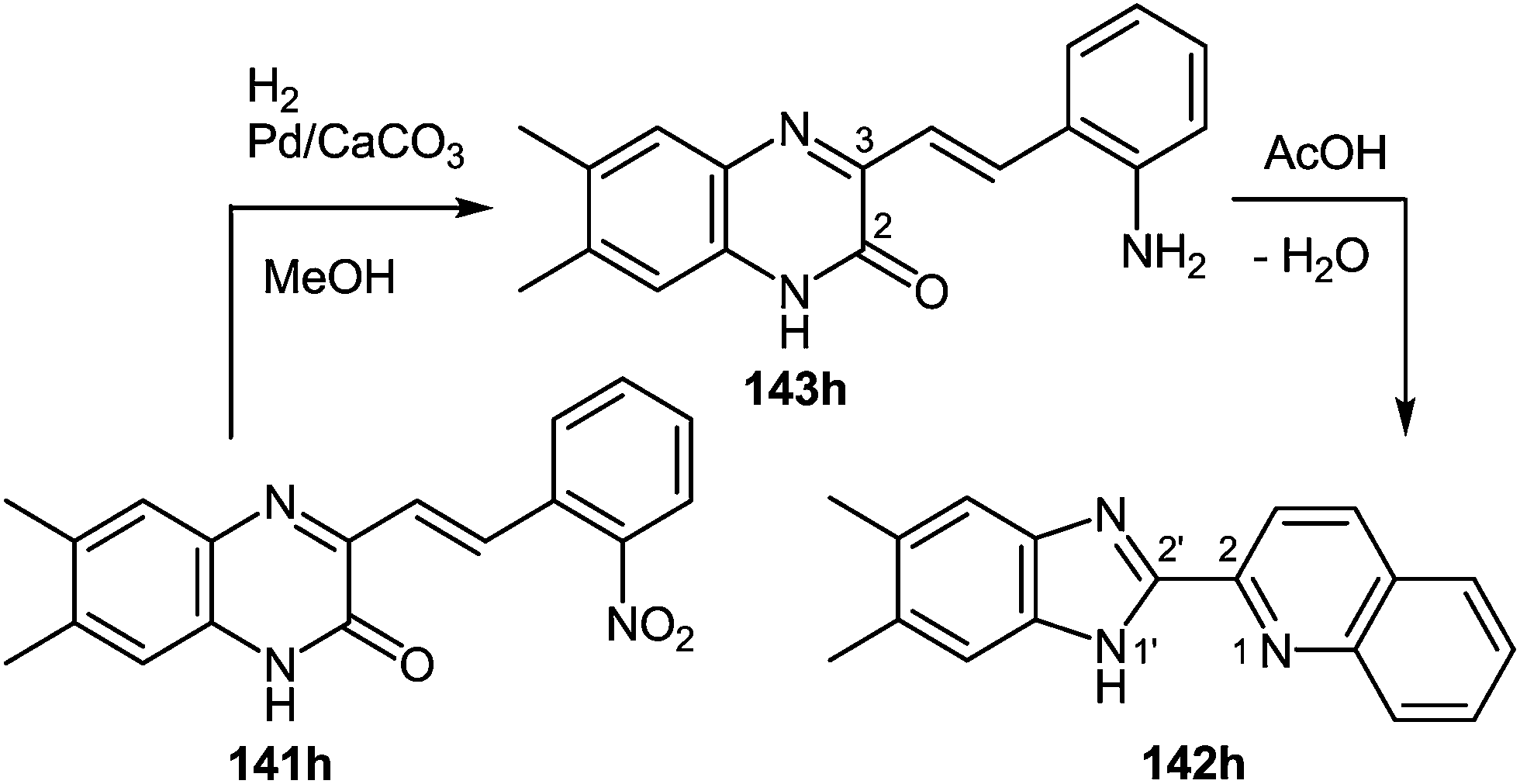 | ||
| Scheme 59 Synthesis of 2-(benzimidazol-2-yl)quinoline from 3-(β-2-nitrostyryl)-6,7-dimethylquinoxalin-2(1H)-one via corresponding amino derivative. | ||
The known chemistry of aniline,114 azadienes,115 and quinoxalinones,105,116 allows to assume that the first stage of this reaction involves the nucleophilic attack of the amine group on the C(3) atom of the quinoxalin-2(1H)-one of A to form the spiro-quinoxaline derivative B. Rearrangement of the spiro-quinoxaline is then assumed to occur according to Scheme 60 by cascade reactions involving: (a) acid-catalysed ring-opening with cleavage of the C(3)–N(4) bond in the spiro-compound C leading to the formation of the quinoline derivative D, and (b) intramolecular nucleophilic attack by the amino group on the carbamoyl carbonyl group with the formation of the final product 142 following the elimination of water.
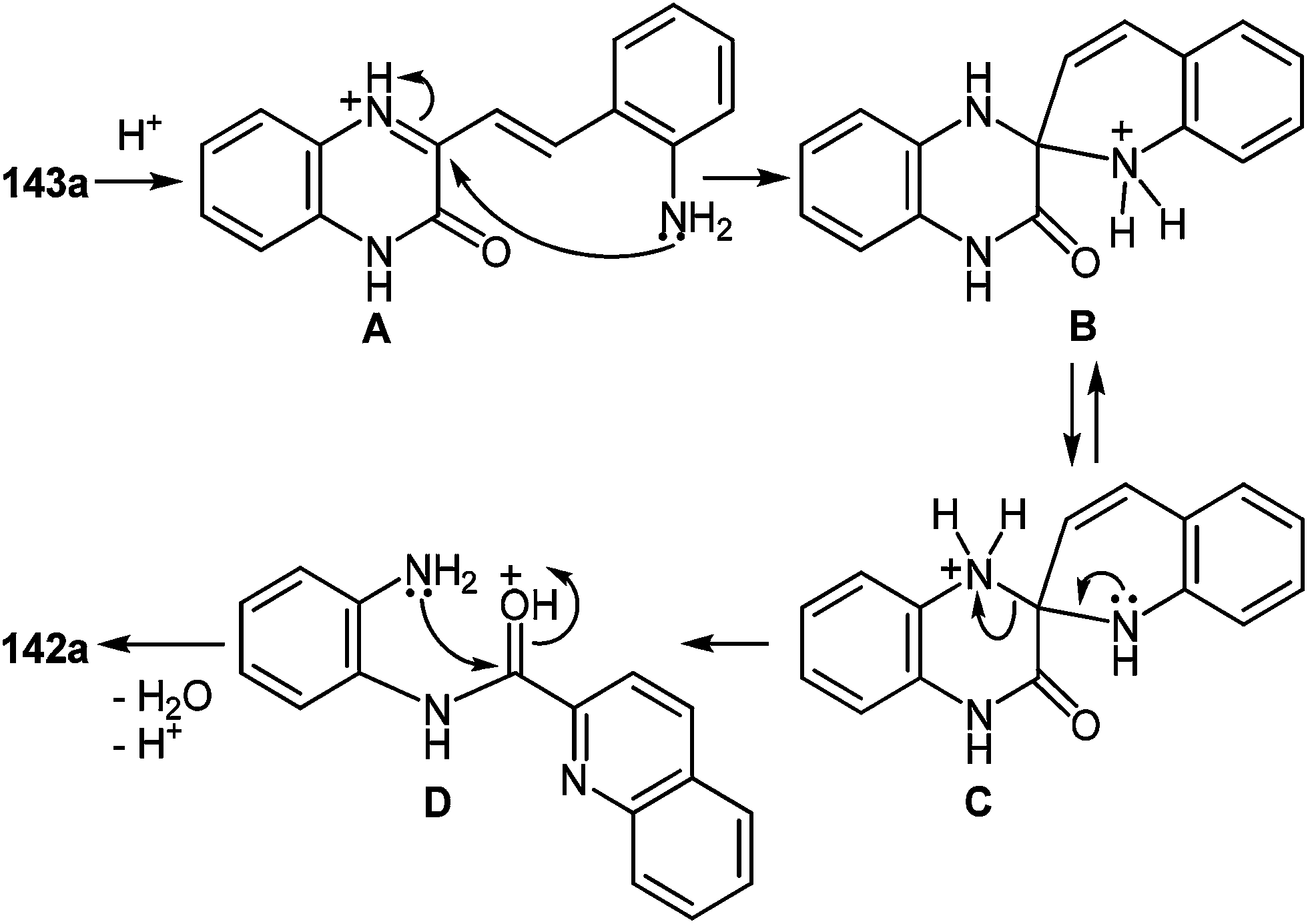 | ||
| Scheme 60 Proposed mechanism of the rearrangement. | ||
This protocol includes a novel acid-catalyzed rearrangement of 3-(β-2-aminostyryl)quinoxalin-2(1H)-ones. The simplicity of the reaction design and the possibility of introducing a variety of substituents at any position of both the benzimidazole and quinoline ring systems makes this method a useful tool for constructing these medicinally and technically (organic emitting materials) relevant compounds. The reaction is readily applicable to large-scale synthesis.
9 Synthesis of 4-(benzimidazol-2-yl)quinolines
3-(β-2-Nitrostyryl)quinoxalin-2-(1H)-ones 141, are easily obtained from 3-methylquinoxalin-2(1H)-one 52 (see Subsection 2.3E) and o-nitrobenzaldehyde 126, and when exposed to sodium dithionite are converted into 2-(benzimidazol-2-yl)quinolines 142.107c The process occurs under reduction conditions in a cascade of the modified Friedländer reaction and the new acid-catalyzed rearrangement through the intermediately formed products – 3-(β-2-aminostyryl)quinoxalin-2(1H)-ones 143 (Scheme 59).The structure of the compound 142 shows that the o-aminostyryle substituent and the C(2), C(3) atoms of the amide and imine fragments of the pyrazine ring of quinoxalin-2(1H)-one 143 (Scheme 59) are involved in the construction of two new heterocyclic systems. In this case, the formation of the pyridine ring of the quinoline system occurs as a result of the proceeding new rearrangement.96,117,118 As seen from the Table 16, the atoms C(2) and C(3) of the quinoxalin-2(1H)-one system become the atoms C(2′) and C(2) of the benzimidazole and quinoline systems of the compounds 142 respectively.
Following the line of logics demonstrated in the papers62c–f,76,102,103,107,108a and specifically in the paper107c on the new rearrangement of the quinoxalin-2(1H)-one derivatives, it was proposed that in the latter reaction 3-(2-aminophenyl)quinoxalin-2(1H)-one derivatives 53 can be used instead of 3-methylquinoxalin-2(1H)-one 52 and o-nitrobenzaldehyde 126 derivatives as the hetero analogues of o-amino aromatic aldehydes and ketones bearing an active α-methylene functionality. The ketones can be condensed with compounds 53 capable of providing a two-carbon fragment in the construction of the quinoline system, which makes it possible to synthesize the 4-(benzimidazol-2-yl)quinolines 145,147,149 isomeric to the 2-(benzimidazol-2-yl)quinolines 142.
The reaction of 3-(2-aminophenyl)quinoxalin-2(1H)-one 53a with acetone in acetic acid proceeded with the formation of the ∼88% yield of the crude product, which contains ∼90% of the quinoline 145a and ∼10% of the quinoxaline 144a derivatives as a result of the rearrangement and as a result of the intramolecular cyclocondensation of the quinoxaline derivative 53a (Scheme 61).119,120
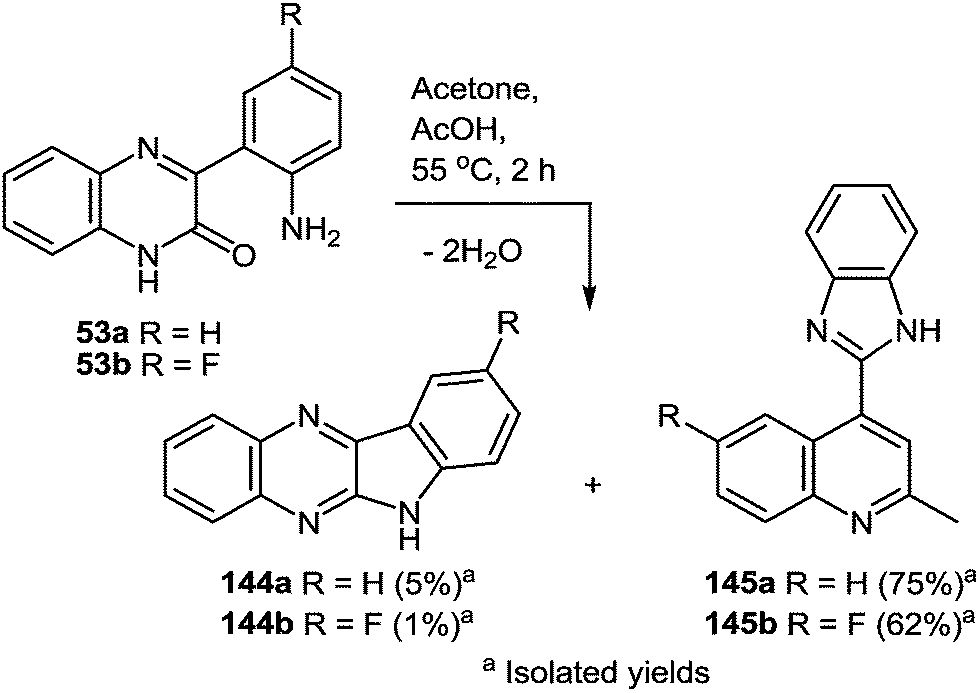 | ||
| Scheme 61 The reaction of 3-(2-aminophenyl)quinoxalin-2(1H)-ones derivatives 53a, b with acetone. | ||
The procedure was extended to 3-(2-amino-5-fluorophenyl)quinoxalin-2(1H)-one 53b and various acetophenones 146a–f. The reactions proceeded very efficiently, and led to the formation of the corresponding 4-(benzimidazol-2-yl)quinolines 147a–l as major and 6H-indolo[2,3-b]quinoxaline 144a–b as minor products (Table 17).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 53 | R | 146 | Ar | Products | Yield (%) | |
| 144 | 147 | ||||||
| 1 | 53a | H | 146a | Ph | 144a + 147a | 6 | 80 |
| 2 | 53a | H | 146b | C6H4Br-4 | 144a + 147b | 4 | 76 |
| 3 | 53a | H | 146c | C6H4Br-3 | 144a + 147c | 4 | 78 |
| 4 | 53a | H | 146d | C6H4Br-2 | 144a + 147d | 4 | 73 |
| 5 | 53a | H | 146e | C6H4Cl-4 | 144a + 147e | 4 | 77 |
| 6 | 53a | H | 146f | C6H4Cl-2 | 144a + 147f | 4 | 71 |
| 7 | 53b | F | 146a | Ph | 144b + 147g | 1 | 68 |
| 8 | 53b | F | 146b | C6H4Br-4 | 144b + 147h | 1 | 66 |
| 9 | 53b | F | 146c | C6H4Br-3 | 144b + 147i | 1 | 64 |
| 10 | 53b | F | 146d | C6H4Br-2 | 144b + 147j | 1 | 61 |
| 11 | 53b | F | 146e | C6H4Cl-4 | 144b + 147k | 2 | 65 |
| 12 | 53b | F | 146f | C6H4Cl-2 | 144b + 147l | 1 | 62 |
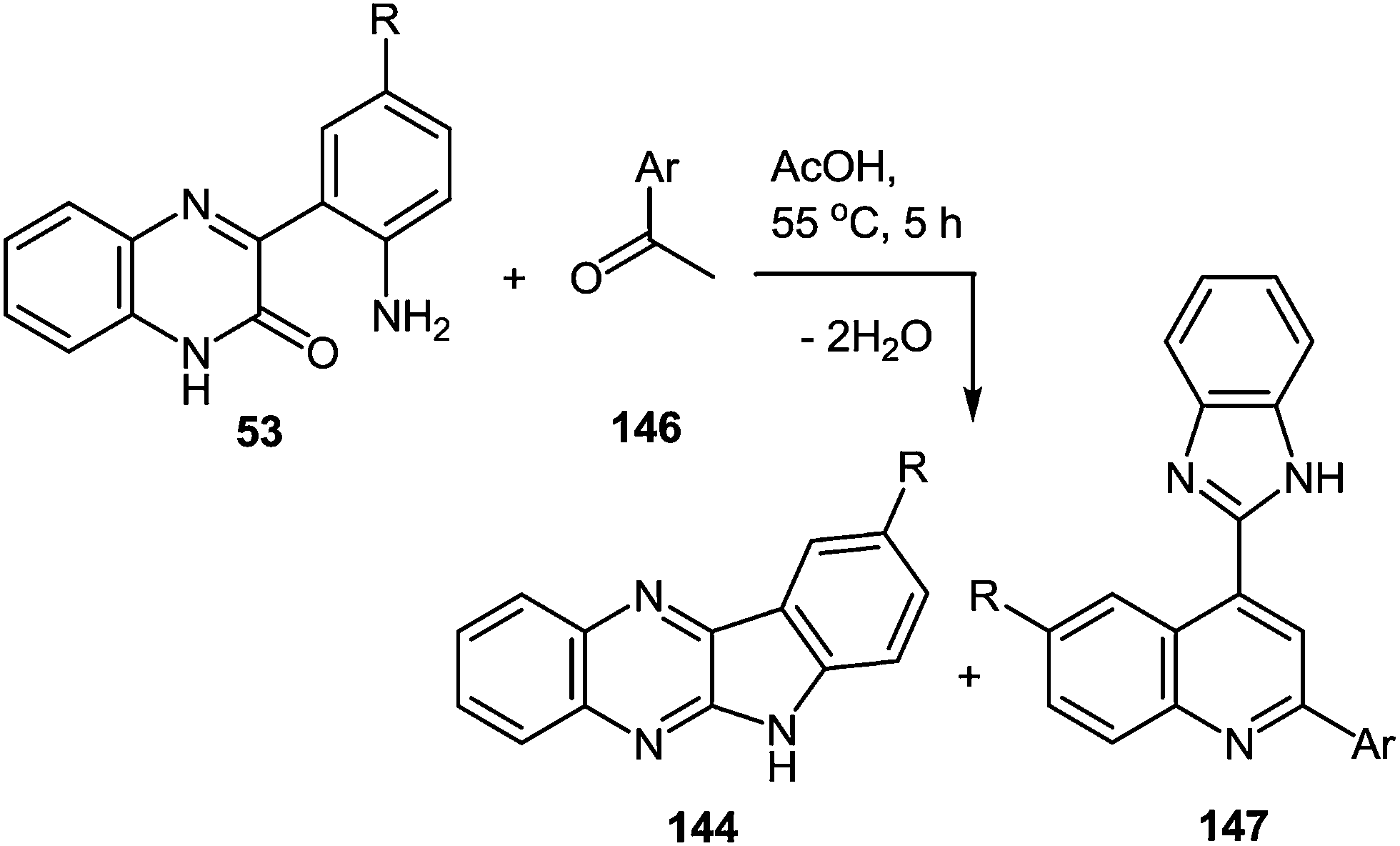
Based on the known chemistry of amines,121 enolizable ketones,122 enamines,123 quinoxalinones,105,116 and the previous reports62e,f,102,103,107,108a a plausible mechanism for the reaction of the formation of 4-(benzimidazol-2-yl)quinolines 147 has been proposed (Scheme 62). The reaction starts with the condensation of ketones with 3-(2-aminophenyl)quinoxalin-2(1H)-one 53a to form imine A, which is transformed to intermediate B by tautomerization. Subsequently intermediate B is easily cyclized through the intramolecular nucleophilic addition to give the spiro-quinoxaline derivative C. The rearrangement of the spiro-quinoxalinone C is then assumed to occur according to Scheme 64, which proceeds by cascade reactions involving: (a) the ring-opening with the cleavage of the C(3)–N(4) bond in the spiro-compound D with the intermediate formation of the quinoline derivative E, (b) the intramolecular nucleophilic attack by the amino group on the carbonyl group with the intermediate formation of the hydroxyl-derivative F, and (c) the elimination of water leading to the formation of the final product 147. All the stages of the reaction involve acid-catalyzed processes.
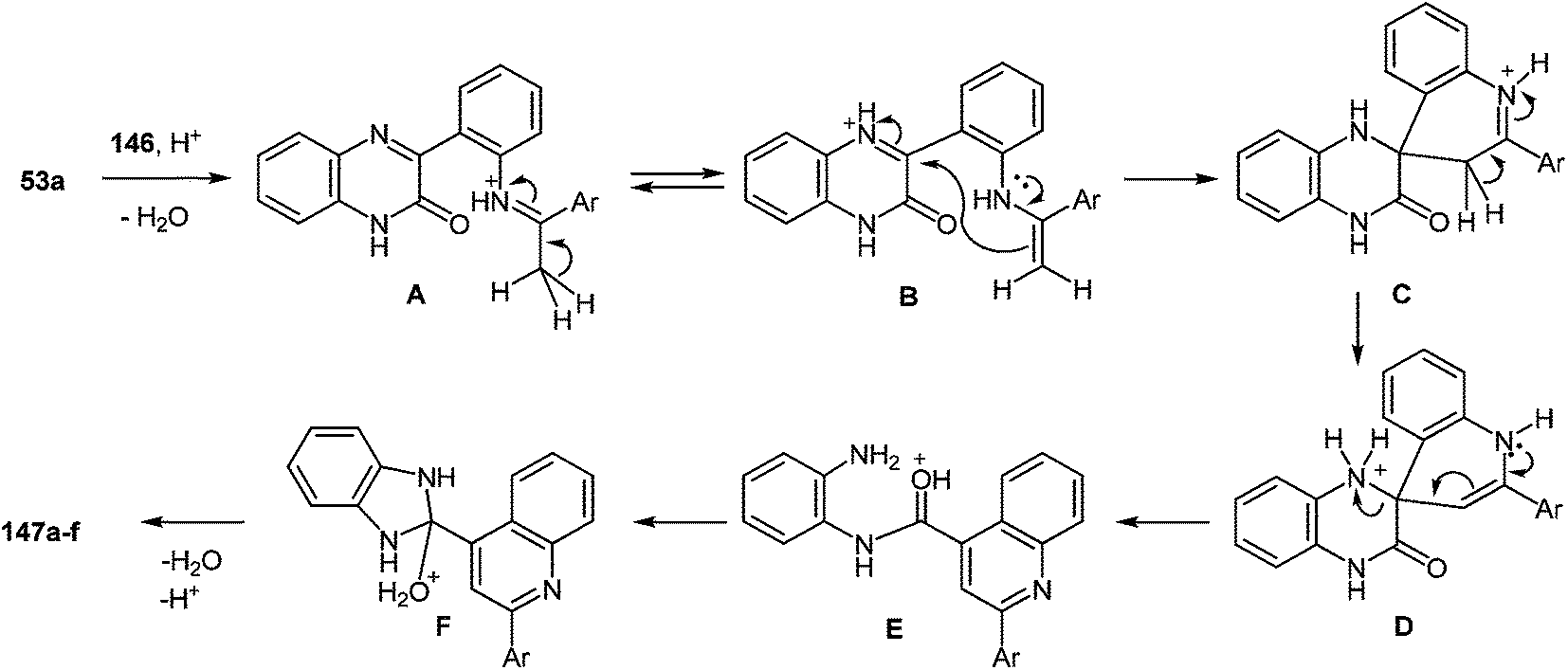 | ||
| Scheme 62 A plausible mechanism for the formation of 4-(benzimidazol-2-yl)quinolines 147. | ||
This chemistry is not limited to mono- and disubstituted systems, and a compound with two acetyl fragments, namely 1,3-diacetylbenzene 148 is an acceptable substrate as well (Scheme 63).
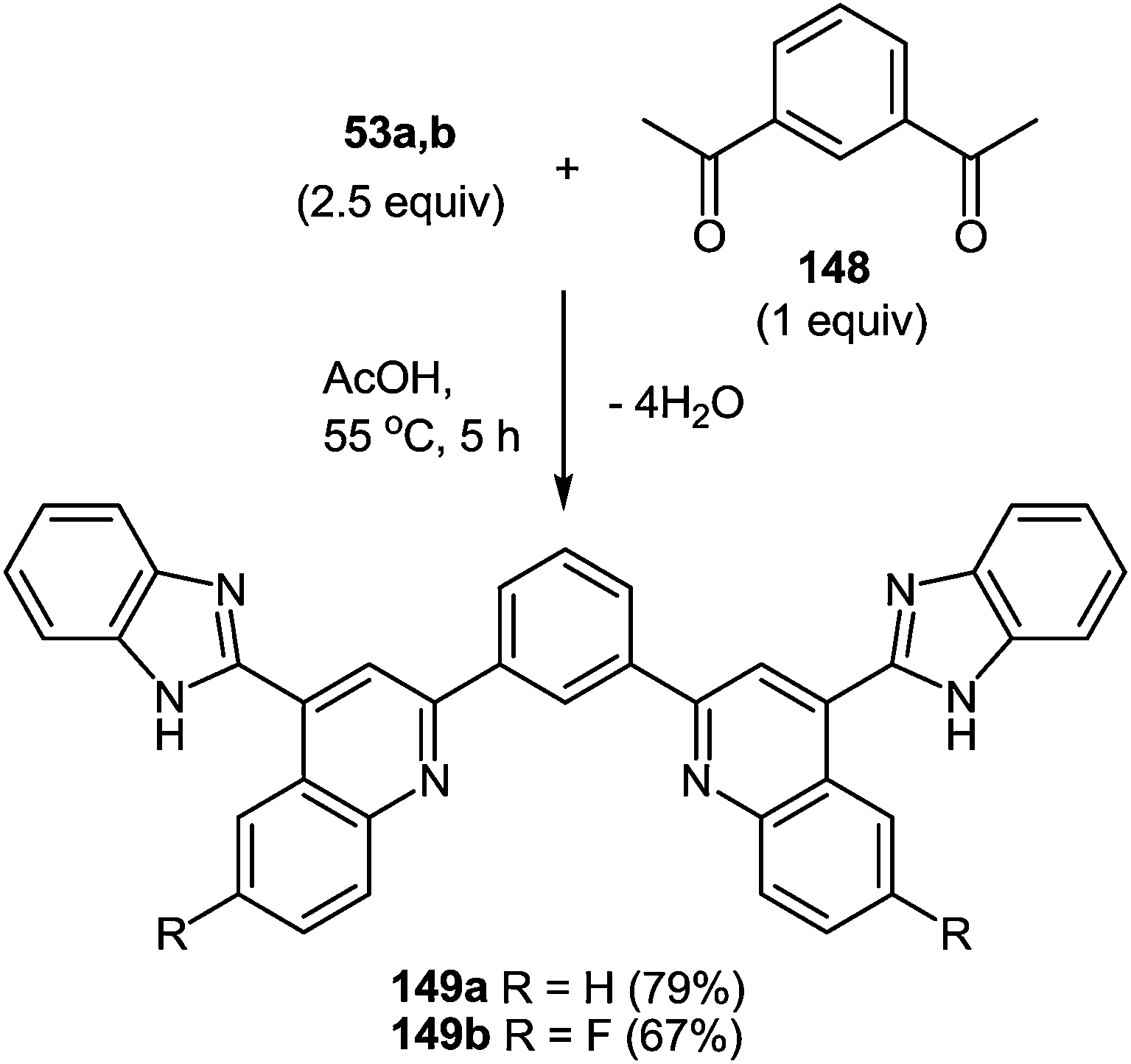 | ||
| Scheme 63 Reaction of 3-(2-aminophenyl)quinoxalin-2(1H)-ones 53a, b with 1,3-diacetylbenzene 148. | ||
The scope of this chemistry was also expanded on the compounds with two same and two different carbonyl groups in their compositions. The 3-(2-aminophenyl)quinoxalin-2(1H)-ones 53a, b were allowed to react with 1,3-pentanedione 150 and ethyl acetoacetate 138, in AcOH, but in these cases with acetyl and ester groups at position 3 of the quinoline system there occurs the formation of benzimidazolo[2,1-a]pyrrolo[3,4-c]quinoline derivatives 153a,b and 154a,b instead of the expected 4-(benzimidazol-2-yl)quinolines 151a,b and 152a,b correspondingly (Scheme 64).102
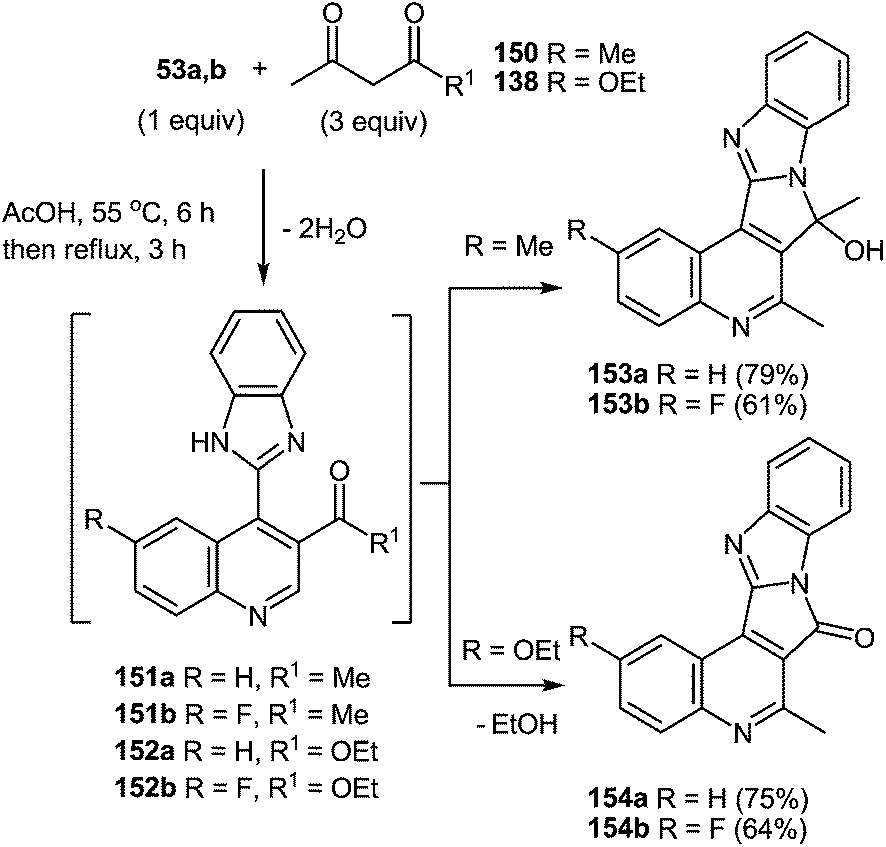 | ||
| Scheme 64 Reaction of 3-(2-aminophenyl)quinoxalin-2(1H)-ones 53a, b with acetylacetone 150 and ethyl acetoacetate 138. | ||
Thus, an efficient synthesis for structurally diverse 4-(benzimidazol-2-yl)quinolines via reactions of 3-(2-aminophenyl)quinoxalin-2(1H)-ones and ketones, including acetone, acetophenones, 1,3-pentanedione and ethyl acetoacetate has been developed. The selective formation of the very different quinoline derivatives depends on the structure of ketones. The key steps are assumed to involve the new acid-catalyzed rearrangement of the spiro-quinoxalinone derivatives formed in situ from the reaction of 3-(2- aminophenyl)quinoxalin-2(1H)-ones and ketones under the modified Friedländer reaction.
It should be pointed out that in an attempt to prepare 3-(2-hydrazinophenyl)quinoxalin-2(1H)-one 157 by reduction of corresponding diazonium salt from 3-(2-aminophenyl)quinoxalin-2(1H)-ones 53 with sodium sulfite instead of the intended product 157 a compound with the molecular formula C14H10N4O has been isolated, which the authors124 considered to be 6H-quinoxalino[1,6-c][1,2,3]benzotriazin-13(12H)-one 158. However, later125 it proved that the structure proposed by the authors124 was incorrect. By NMR spectroscopy125 and by X-ray analysis126 it was shown that the correct structure is 1-(1H-indazol-3-yl)-1H-benzimidazol-2(3H)-one 155 (Scheme 65). The formation of the 1H-benzimidazol-2(3H)-one derivative 155 as a consequence of the principle suggested above proceeds through the spiro-compound 156 without any mobile hydrogen atom in their spiro-forming component.
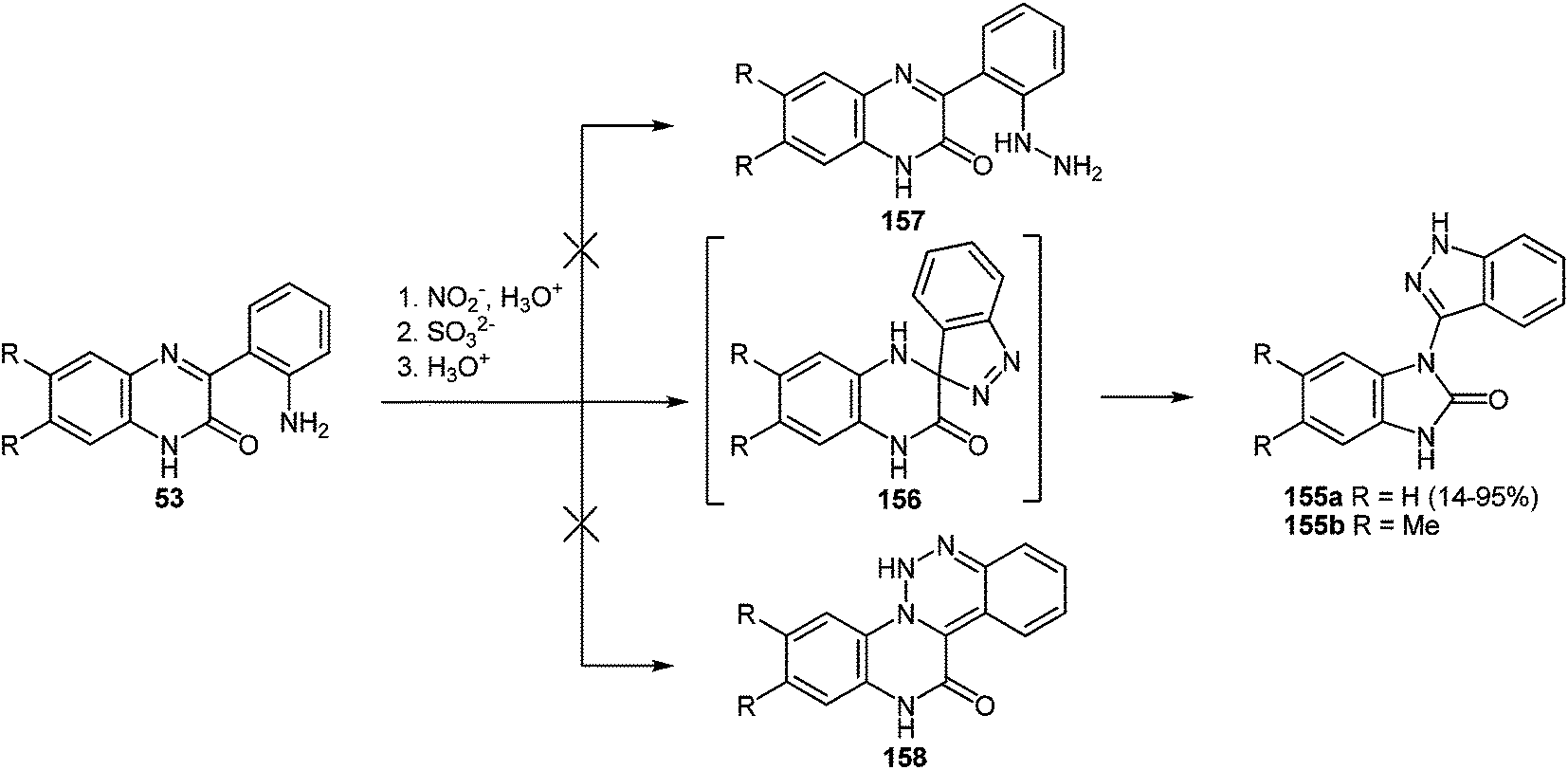 | ||
| Scheme 65 Reaction of 3-(2-aminophenyl)quinoxalin-2(1H)-ones 53 with NaNO2 in acidic medium. | ||
10 Synthesis of 1-pyrrolylbenzimidazolones and their aza-analogues via new rearrangements
Scheme 16 shows that the key step of the reactions above, involved a novel acid-catalyzed rearrangement of intermediate spiro-quinoxalin-2(1H)-one derivatives with a contraction of the pyrazine ring of the quinoxalin-2-one system. It was also shown that the necessary condition for the rearrangement is the presence of at least one mobile hydrogen atom in the spiro-forming fragment, which is responsible for the elimination of water. As can be seen from Scheme 16 the formation of water takes place with the involvement of the oxygen atom of the carbonyl group. We assumed that if the spiro-quinoxalinone derivative with no mobile hydrogen atom in the spiro-fragment was subjected to rearrangement, there might probably be two options. The first one is that the rearrangement would not occur at all, the second one is that another rearrangement would take place without any water elimination and with the preservation of the carbonyl oxygen atom, probably quinoxalinone-benzimidazolone. To confirm this assumption it was necessary to synthesize a spiro-derivative so the quinoxalinone without any mobile hydrogen atoms. To this end, after analyzing all the possible nucleophilic reagents, we have chosen the enamines as CN-nucleophiles. The reaction of 3-aroyl- and alkanoylquinoxalin-2(1H)-ones with the commercially available enamines (methyl- (159a) and ethyl- (159b) 3-aminocrotonates) as CN-nucleophiles under the acid catalysis condition proceeds so fast that it appears impossible to allocate the expected spiro-compound which are immediately subjected to the rearrangement with the formation of the corresponding N-(pyrrol-3-yl)benzimidazol-2-ones (160) as major and N-(pyrrol-2-yl)benzimidazol-2-ones (161) as minor products with the overall 89–99% yields (Table 18).127|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | 37 | R1 | R2 | R3 | 159 | R4 | Products (yield) | Overall yield (160/161)a |
| a Ratio determined by the 1H NMR of the crude products. | ||||||||
| 1 | 37a | H | H | Ph | 159a | Me | 160a + 161a | 97%, 78:22 |
| (45%) (n/i) | ||||||||
| 2 | 37b | H | H | C6H4F-4 | 159a | Me | 160b + 161b | 94%, 74:26 |
| (42%) (n/i) | ||||||||
| 3 | 37c | H | H | C6H4Cl-4 | 159a | Me | 160c + 161c | 97%, 72:28 |
| (52%) (n/i) | ||||||||
| 4 | 37d | H | H | C6H4Br-4 | 159a | Me | 160d + 161d | 97%, 70:30 |
| (51%) (n/i) | ||||||||
| 5 | 37e | H | H | C6H4I-4 | 159a | Me | 160e + 161e | 98%, 75:25 |
| (53%) (n/i) | ||||||||
| 6 | 37i | H | H | n-Pr | 159a | Me | 160f + 161f | 97%, 91:9 |
| (39%) (n/i) | ||||||||
| 7 | 37x | Me | Me | Ph | 159a | Me | 160g + 161g | 97%, 83:17 |
| (62%) (n/i) | ||||||||
| 8 | 37t | COPh | H | Ph | 159a | Me | 160h + 161h | 97%, 79:21 |
| (43%) (n/i) | ||||||||
| 9 | 37a | H | H | Ph | 159b | Et | 170i + 171i | 99%, 78:22 |
| (58%, 24%) (4%) | ||||||||
| 10 | 37b | H | H | C6H4F-4 | 159b | Et | 160j + 161j | 96%, 77:23 |
| (59%) (n/i) | ||||||||
| 11 | 37c | H | H | C6H4Cl-4 | 159b | Et | 160k + 161k | 97%, 67:33 |
| (61%, 40%) (5%) | ||||||||
| 12 | 37d | H | H | C6H4Br-4 | 159b | Et | 160l + 161l | 99%, 67:33 |
| (56%, 38%) (4%) | ||||||||
| 13 | 37e | H | H | C6H4I-4 | 159b | Et | 160m + 161m | 98%, 60:40 |
| (56%) (n/i) | ||||||||
| 14 | 37x | Me | Me | Ph | 159b | Et | 160n + 161n | 99%, 84:16 |
| (65%) (n/i) | ||||||||
| 15 | 37t | COPh | H | Ph | 159b | Et | 160o + 161o | 98%, 72:28 |
| (41%) (n/i) | ||||||||
| 16 | 37u | CO2H | H | Ph | 159b | Et | 160p + 161p | 89%, 75:25, (inseparable mixture) |
| (77%) | ||||||||
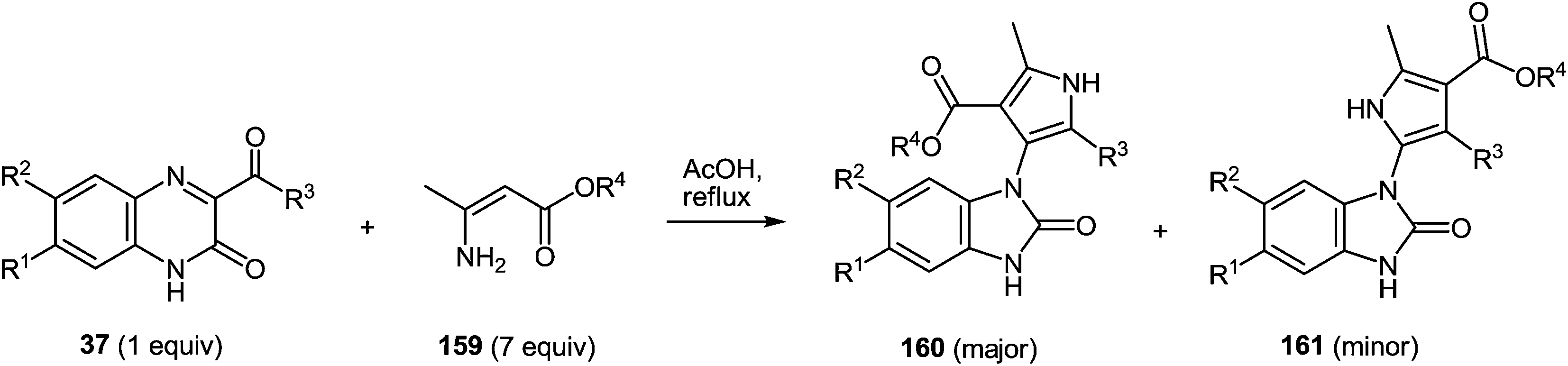
A plausible reaction mechanism for the formation of N-pyrrolylbenzimidazol-2-ones 160 and 161 has been proposed (Scheme 66). The formation of N-pyrrolylbenzimidazol-2-ones occurs in two different ways (pathway I and pathway II), differing at their initial stage of the process. In the case of the formation of N-(pyrrol-3-yl)benzimidazol-2-ones 160 the reaction starts (Scheme 68) with the acid catalyzed subsequent Michael type of the reaction128 between 37a and 159a involving a nucleophilic attack by the enamino double bond (of 159a) on the electron deficient double bond (of 37a), which leads to the formation of A. The intramolecular cyclization of A involving the attack by the imine nitrogen on the nearby –C(O)Ph moiety affords the spiro-quinoxaline derivative B. The rearrangement of the spiro-quinoxalinone B is then assumed to occur according to Scheme 68, which proceeds by cascade reactions involving: (a) the intramolecular nucleophilic attack by the amino group on the carbonyl group with the intermediate formation of the hydroxy-derivative C, (b) the ring-opening with the cleavage of the C(2)–C(3) bond in the hydroxy-derivative C with the elimination of water leading to the formation of the final product 160a.
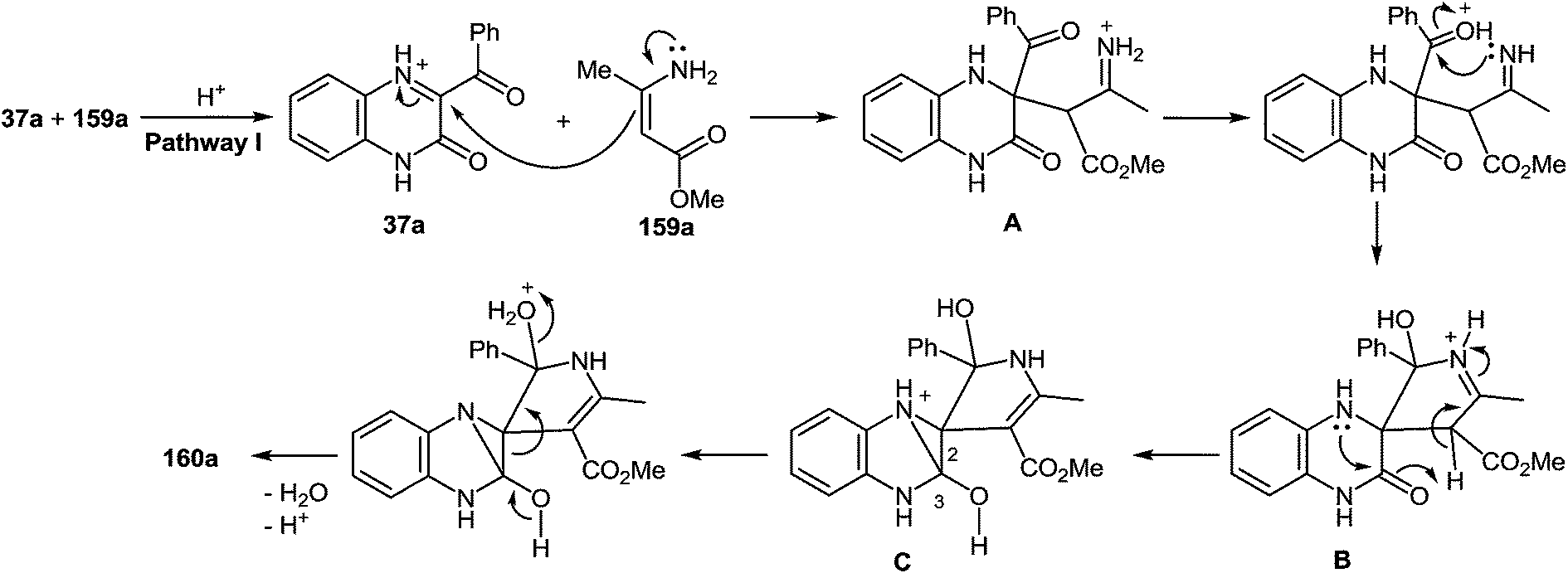 | ||
| Scheme 66 Proposed mechanisms for the formation of 160a (pathway I – via an initial attack on the C(3) atom of quinoxalin-2(1H)-one). | ||
In the case of the formation of N-(pyrrol-2-yl)benzimidazol-2-ones 161 at its initial stage there occurs a nucleophilic attack by the enamino double bond (of 159a) on the electron deficient benzoyl carbonyl group (of 37a) which leads to the formation of D (Scheme 68, pathway II). This brings about the rearrangement product via intermediates E and F (Scheme 67).
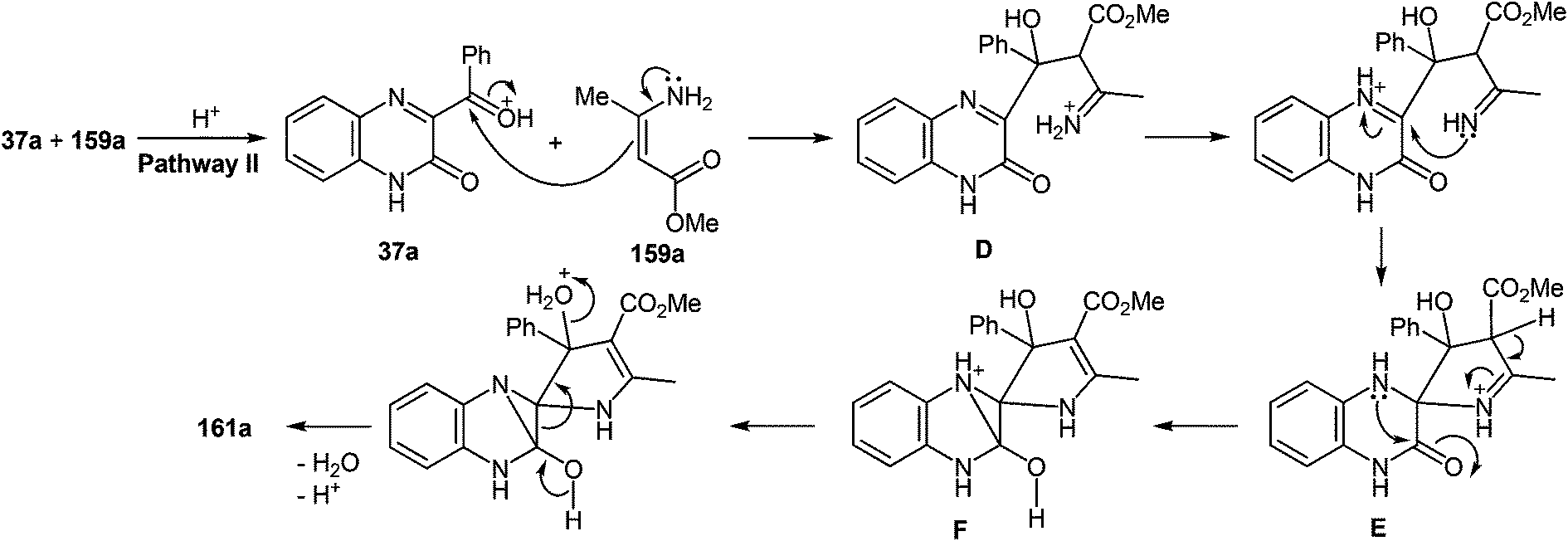 | ||
| Scheme 67 Proposed mechanisms for the formation of 161a (pathway II – via an initial attack on the C atom of benzoyl group). | ||
This chemistry is not limited to the quinoxalin-2(1H)-ones and the 5- and 7-aza-quinoxalin-2(1H)-ones, namely 3-benzoylpyrido[3,2-b]pyrazin-2(1H)-one 95a and 3-benzoylpyrido[3,4-b]pyrazin-2(1H)-one 95d subjected to the rearrangement with the formation easily separable regioisomeric products 162/163 and 164/165 with overall quantitative yields (Scheme 68).129
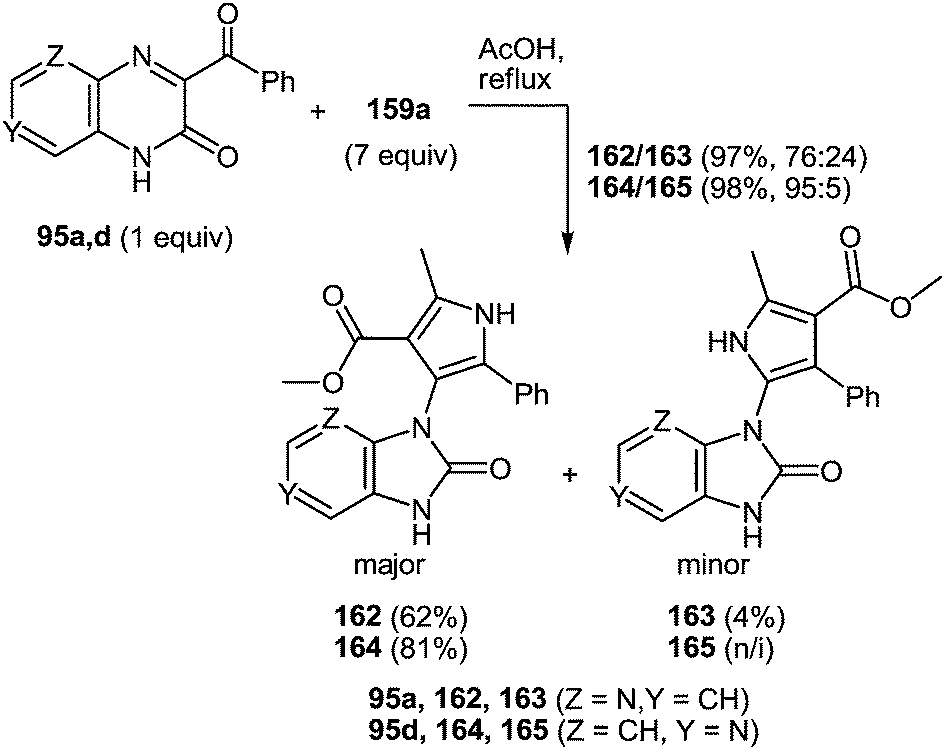 | ||
| Scheme 68 Synthesis of N-pyrrolyl-1H-imidazo[5,4-b]- (162/163) and N-pyrrolyl-1H-imidazo[4,5-c]pyridin-2(3H)-ones (164/165). | ||
In comparison with the existing methods, the present approach offers the following advantages: (i) it proceeds faster and affords good to excellent yields under mild conditions with no additional activation modes such as microwave irradiation, (ii) it is very cost-effective and uses the inexpensive easily and commercially available reagents, and (iii) it is applicable to a broader range of substrates, including 3-aroyl(alkanoyl)quinoxalin-2(1H)-ones, 3-benzoylpyrido[3,2-b]pyrazin-2(1H)-one and 3-benzoylpyrido[3,4-b]pyrazin-2(1H)-one and various enamines.
An effective synthesis strategy has been developed via the novel quinoxalin-2(1H)-one/benzimidazol-2-one rearrangement that makes possible a rapid access to the N-pyrrolylbenzimidazol-2-ones from the readily available 3-aroylquinoxalin-2(1H)-ones with various substituents and commercially available methyl and ethyl-3-aminocrotonates. The methodology was found to be general and a wide variety of N-pyrrolylbenzimidazol-2-one derivatives were prepared in good yields. Due to the availability of the starting materials and the potential applications of the products, this method is highly perspective in organic synthesis and medicinal chemistry.
If instead of commercially available enamines the heterocyclic compound with an enamine fragment is used the rearrangement found allows to synthesize more complex heterocyclic system that are hard-to-reach by other known methods. Indeed, carrying out the reaction of 3-(benzimidazo-2-yl)quinoxalin-2(1H)-one 37s with 3-(benzimidazol-2-yl)methylenequinoxalin-2(1H)-one 107 in boiling AcOH led to the derivative of pyrrolo[1,2-a]quinoxalin-4(5H)-one 166 in a good yield (Scheme 69).102
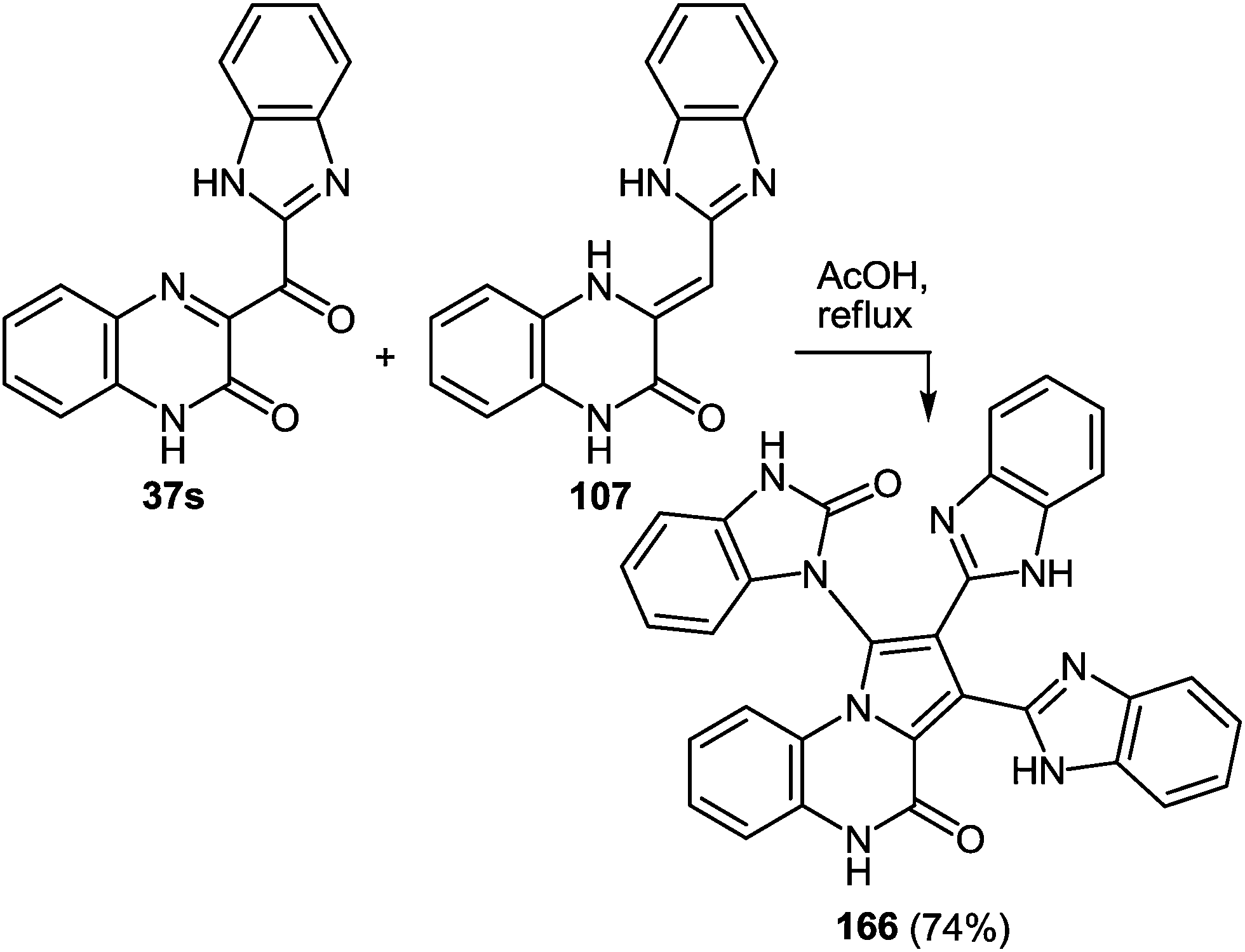 | ||
| Scheme 69 The formation of 1-(benzimidazol-2-on-1-yl)-2,3-(dibenzimidazol-2-yl)pyrrolo[1,2-a]quinoxalin-4(5H)-one 166. | ||
The formation of compound 166 occurs as a result of a cascade reaction involving: (a) the intermolecular ene reaction130 between the enamine 107 and ketone 37s with the formation of compound G, (b) the intramolecular ene reaction131 with the formation of the spiro-derivative quinoxalinono[2′,1]pyrrolo[1,2-a]quinoxaline H, and (c) a new quinoxalinono-benzimidazolono rearrangement in the spiro-forming fragment. Unlike the rearrangements discussed above (see Sections 2.2 and 4–9), in which the opening of the pyrazine ring of the quinoxalinone system of spiro-derivative (Schemes 14, 46, 47, 51, 52, 58, 62 and 64) occurs between the C(3) and N(4) atoms, the opening of the pyrazine ring of the quinoxaline system of spiro-derivative H occurs in the new rearrangement between the C(2) and C(3) atoms, through the intermediate formation of fused spiro-aziridino[2′,1]pyrrolo[1,2-a]quinoxaline I (Scheme 70).
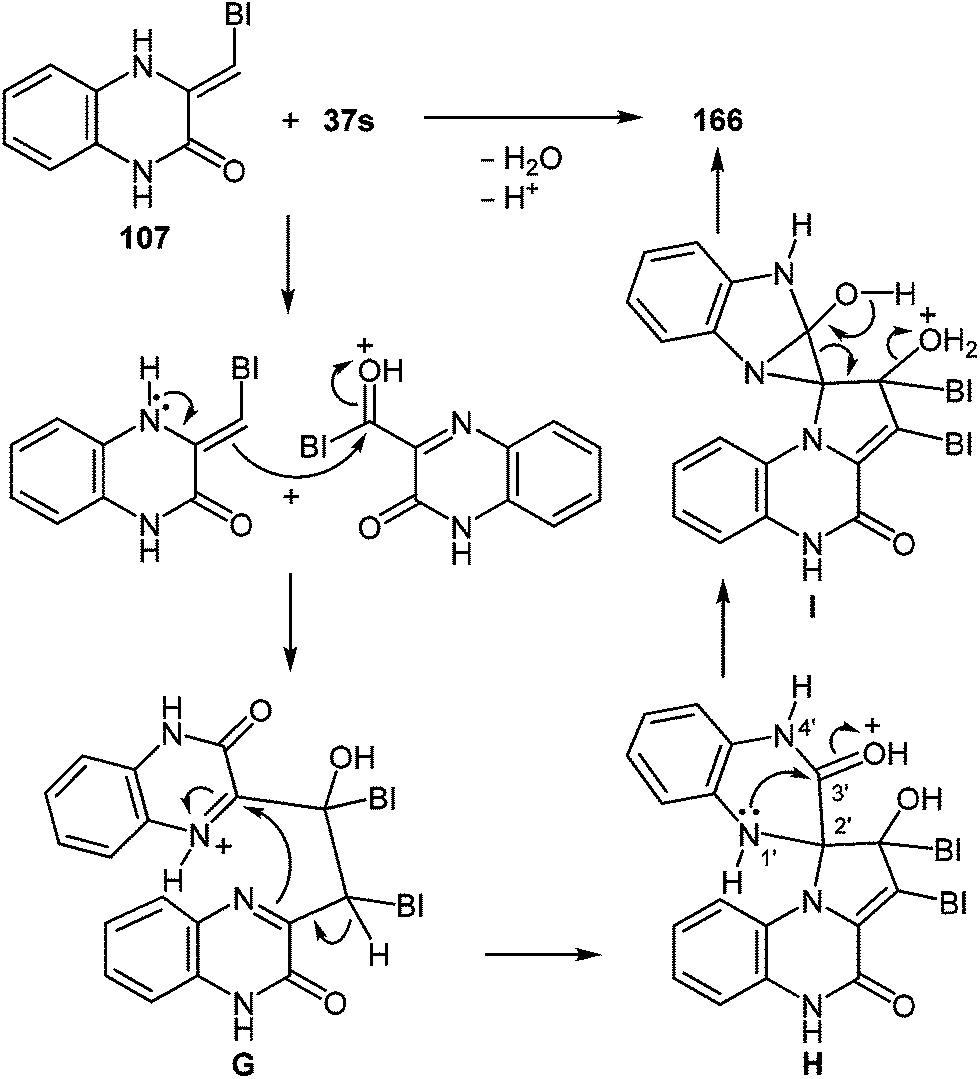 | ||
| Scheme 70 A plausible mechanism for the formation of pyrrolo[1,2-a]quinoxalin-4(5H)-one 166. | ||
In this case 3-(benzimidazo-2-yl)quinoxalin-2(1H)-one 37s, supplying two carbon atoms in the formation of the pyrrole ring is subjected to a rearrangement (Scheme 69) different from that discussed above (see Section 4.2).
Enamines for the rearrangement can be generated from decomposition of azides. The above rearrangement was observed earlier in our group on the example of the self-condensation of 3-(α-azidophenylalkyl)quinoxalin-2(1H)-ones 167a–c (Scheme 71) (see D. F. Saifina thesis pages 89–93 in chapter 4).89 In this version of the rearrangement the enamines for the rearrangement can be generated in situ by the decomposition of azides. The final products 168a–c are formed as a result of the intermolecular condensation of an enamine J and the corresponding ketone K formed in situ under the reaction conditions. In this case the new rearrangement of quinoxalinones makes it possible to simultaneously construct two various new heterocyclic systems under one-pot reaction conditions.
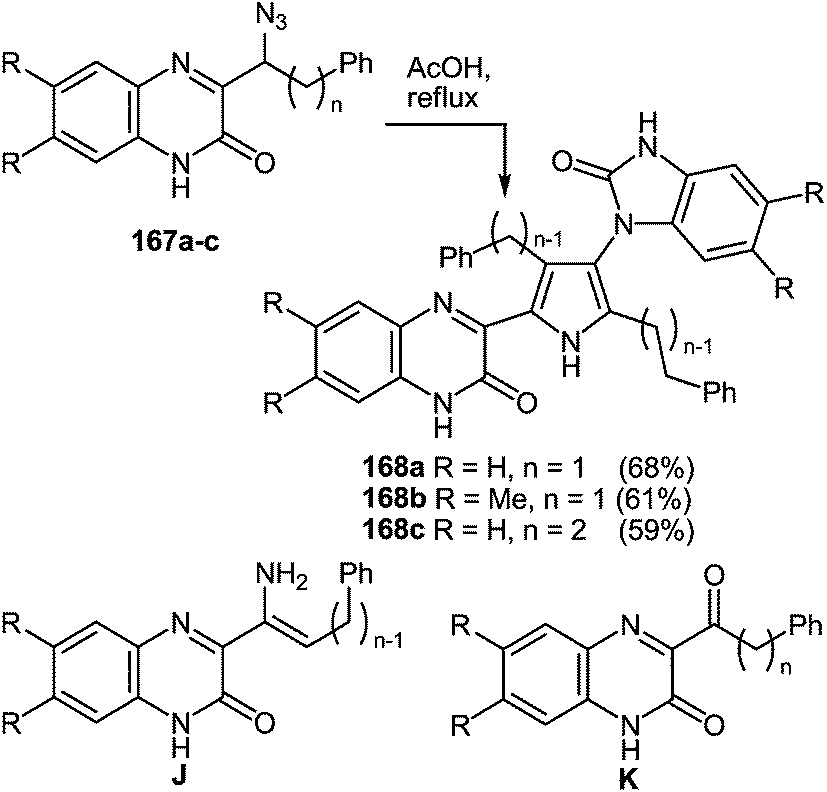 | ||
| Scheme 71 A new quinoxalinone–benzimidazolone rearrangement proceeding under self-condensation of 3-(α-azidophenylalkyl)quinoxalin-2(1H)-ones. | ||
The fact that the formation of the benzimidazolone derivatives in the reactions of the self-condensation of 3-(α-azidophenylalkyl)quinoxalin-2(1H)-ones 167 (Scheme 73) and in the condensation of 3-(benzimidazo-2-yl)quinoxalin-2(1H)-one 37s with its predecessor – 3-(benzimidazol-2-yl)methylenequinoxalin-2(1H)-one 107 (Scheme 71) gives reason for propose a new hypothesis, that “any of the spiro-derivatives of 1,2,3,4-tetrahydroquinoxalin-3-one without any mobile hydrogen atom in their spiro-forming component are on their way to the benzimidazolone derivative with the spiro-forming component at position 1” (Scheme 72).
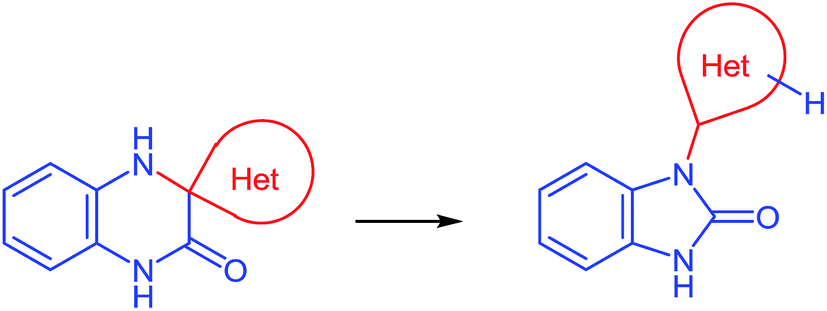 | ||
| Scheme 72 A schematic presentation of the spiro-1,2,3,4-tetrahydroquinoxalin-3-one → benzimidazolone rearrangement. | ||
The above reactions of 3-benzoyl-, 3-alkanoyl- quinoxalinones proceed equally well with enamines generated in situ from the ketones with an activated methylene group and amines.
Table 19 reports the structural variations which are tolerated by these three-component reactions of 3-aroylquinoxalinones with various ketones and ammonium acetate.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | 37 | R1 | R2 | 146 | R3 | Time (h)/NH4OAc (eq.) | Product | Yield (%) |
|
a Formed two isomers in a 63:37 percentage ratio (based on 1H NMR).
b 55% of 37t was recovered.
|
||||||||
| 1 | 37a | H | H | 146a | Ph | 20/20 | 169a | 65 |
| 2 | 37a | H | H | 146b | C6H4Br-4 | 14/15 | 169b | 81 |
| 3 | 37a | H | H | 146c | C6H4Br-3 | 20/20 | 169c | 66 |
| 4 | 37a | H | H | 146d | C6H4Br-2 | 20/20 | 169d | 62 |
| 5 | 37a | H | H | 146e | C6H4Cl-4 | 14/15 | 169e | 79 |
| 6 | 37a | H | H | 146f | C6H4Cl-2 | 20/20 | 169f | 63 |
| 7 | 37a | H | H | 146g | C6H4NO2-3 | 20/20 | 169g | 59 |
| 8 | 37a | H | H | 146h | C6H4OMe-4 | 14/15 | 169h + 170h | 92a |
| 9 | 37a | H | H | 146i | Py-2 | 20/20 | 169i | 79 |
| 10 | 37a | H | H | 146j | Py-3 | 20/20 | 169j | 73 |
| 11 | 37a | H | H | 146k | Py-4 | 20/20 | 169k | 76 |
| 12 | 37u | CO2H | H | 146a | Ph | 14/15 | 169l | 62 |
| 13 | 37u | CO2H | H | 146b | C6H4Br-4 | 14/15 | 169m | 84 |
| 14 | 37t | COPh | H | 146b | C6H4Br-4 | 24/25 | 169n | 12b |
| 15 | 37x | Me | Me | 146b | C6H4Br-4 | 24/25 | 169o | 63 |
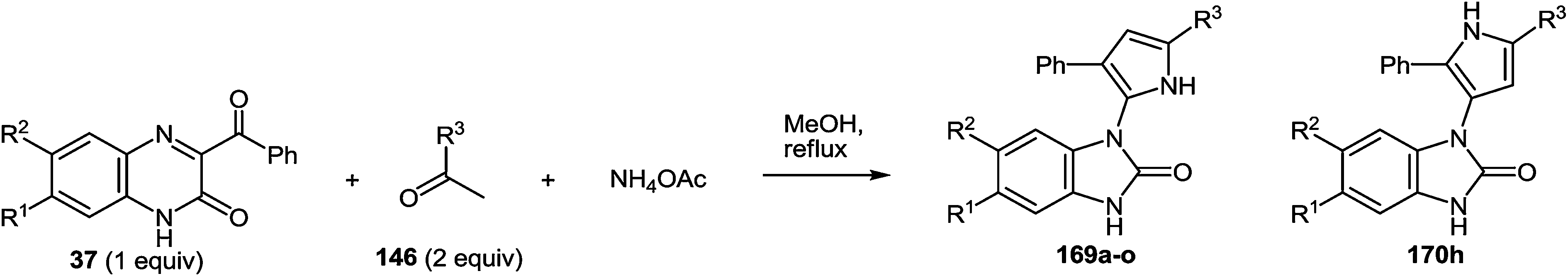
Acetophenones (electron-withdrawing and electron-donating substituents) and heteroaromatic ketones have resulted in the corresponding products in good to excellent yields (Table 19). The same is true for the different 3-BQs with various substituents with the exception of the PhC(O) group (Table 19, entry 14) in the benzene ring of the quinoxalin-2(1H)-one system.
The replacement of the commercially available acetophenone 146a with 1,3-diacetylbenzene 148, bearing an additional acetyl group, would allow the anticipated cascade process with two MCR modifications in one pot with the formation of compound 172, with two 1-(pyrrol-2-yl)benzimidazolone cores in the benzene ring as a major product and compound 171 as a minor product with one 1-(pyrrol-2-yl)benzimidazolone core. It has been shown that acetophenone 171 with 3-BQ 37a in the presence of NH4OAc (the ratio of reagents is given in the Scheme 74) can also be transformed to 172 with a 30% yield (Scheme 73).
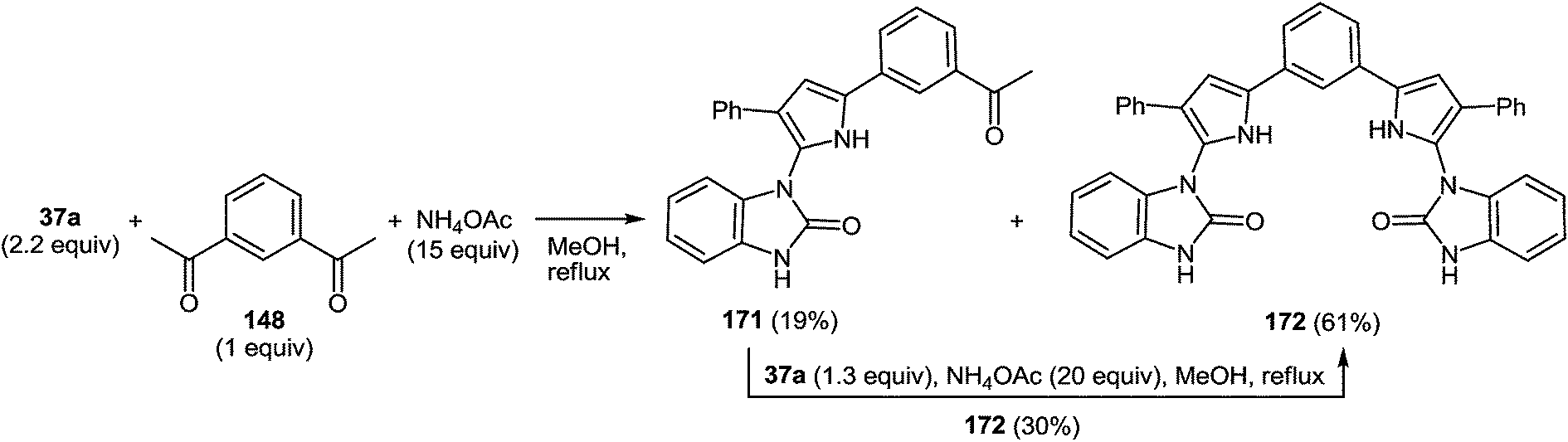 | ||
| Scheme 73 Synthesis of 1-[5-(3-acetylphenyl)-3-phenylpyrrol-2-yl]benzimidazol-2(3H)-one 171 and 1,3-bis[2-(benzimidazol-2-on-1-yl)-3-phenylpyrrol-5-yl]benzene 172. | ||
The known chemistry of ketones,122 imines,121 quinoxalines105 and enamines123 allows to propose a plausible reaction course (Scheme 74), although the exact mechanism of this reaction is yet unclear. Intermediate L formed at the initial stage of the processes reacts with the 3-BQ 37a in two different ways (pathway I and pathway II) with the formation of an isomeric spiro[pyrrol-3,2′-quinoxalin]-3-one derivative O and a spiro[pyrrol-2,2′-quinoxalin]-3-one derivative Q through the intermediate M and N. The latter are formed by the initial attack of enamine on the benzoyl carbonyl carbon atom (pathway I) and on the C(3) atom of the quinoxalinone system (pathway II), respectively. Then, both pathway I and pathway II proceed by cascade reactions involving: (a) the consecutively acid-catalyzed ring-closure of quinoxaline derivatives M, N to spiro-derivatives Q, O followed by formation of intermediates R, P with the aziridine ring system; (b) the acid-catalyzed ring-opening in intermediates R, P with the formation of the final 1-(pyrrol-2-yl)- 169 and 1-(pyrrol-3-yl)benzimidazolone 170 derivatives.
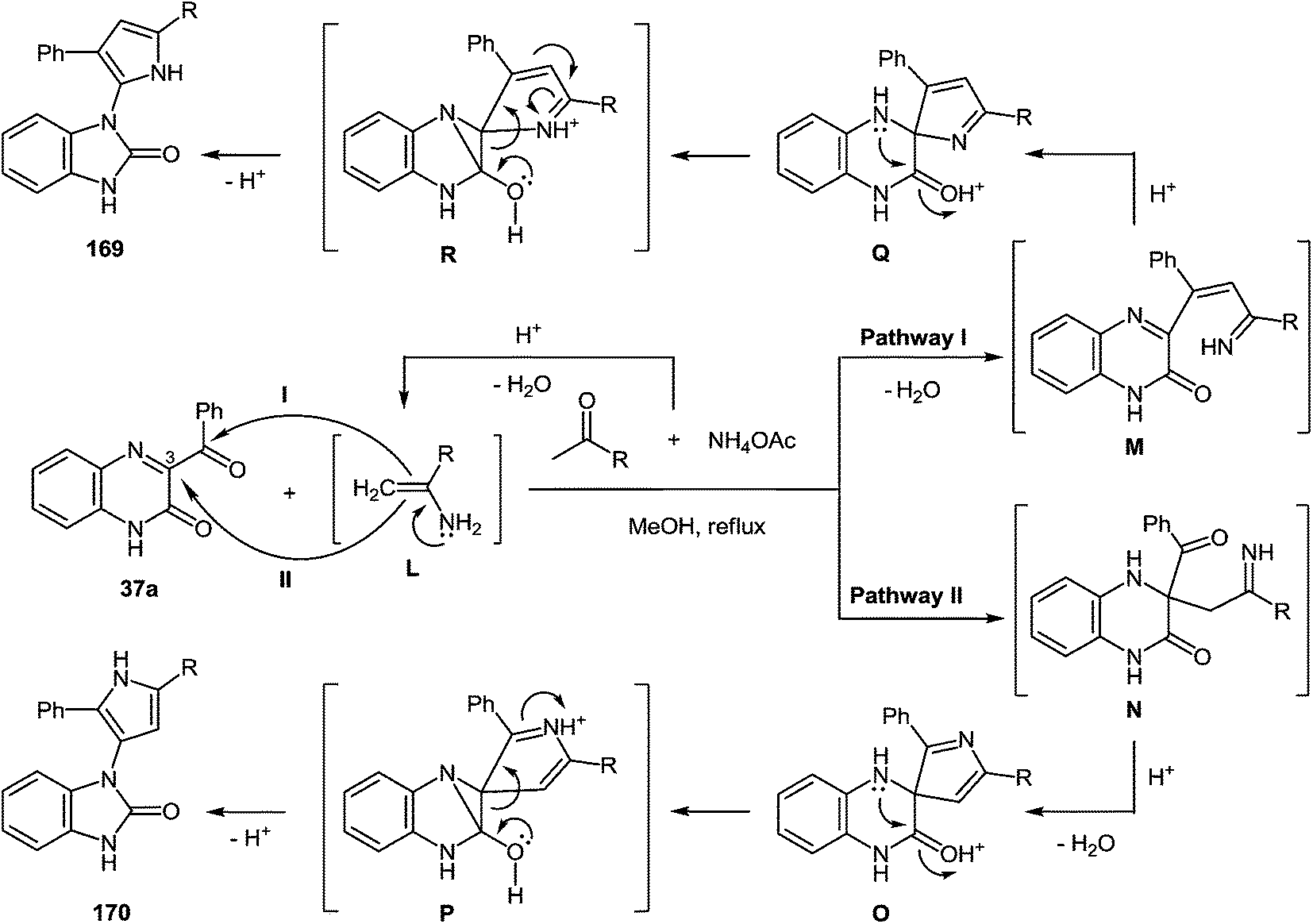 | ||
| Scheme 74 A plausible mechanism for the formation of 1-(5-R-3-phenylpyrrol-2-yl)- (169) and 1-(5-R-2-phenylpyrrol-3-yl)- (170) benzimidazol-2(3H)-ones. | ||
An important three component reaction of 3-benzoylquinoxalinones, various methylaryl-(hetaryl)ketones and ammonia has been developed. The method allows the preparation of substituted 1-(pyrrolyl)benzimidazolone derivatives via multicomponent reaction conditions from easily available 3-benzoylquinoxalinone precursors in the presence of various methylaryl(hetaryl)ketones and ammonia with good to excellent yields. Enamines could be generated in situ from ketones and ammonia, which smoothly react with 3-benzoylquinoxalinones to provide 1-(pyrrolyl)benzimidazolone derivatives. An important aspect of this method is that it can be used for the synthesis of a wide range of benzimidazolone derivatives, since various methylketones are commercially available and can easily be obtained through the various simple reactions.
11 Conclusion
Benzimidazol(on)es have taken a leading role in the recent literature because of their wide variety of applications in various disciplines like medicinal chemistry, organometallics, and material science. Several novel synthetic routes have been developed to achieve these skeletons. These newer synthetic routes are based on the combination of several interesting strategies such as tandem sequences, multicomponent reactions, and rearrangements. As is evident from the discussion in this review, these synthetic procedures offered easy access to benzimidazol(on)es from simple and readily available precursors. The development of these synthetic procedures is very useful especially for medicinal and material chemists. In this review we mainly focused our attention to two fundamentally new rearrangements of quinoxalinones and their aza-analogues discovered by us.The first one can be schematically represented as TYPE 1 on the Scheme 75: “Any of the spiro-derivatives of 1,2,3,4-tetrahydroquinoxalin-3-one with at least one mobile hydrogen atom in their spiro-forming component are on their way to the benzimidazole derivative with the spiro-forming component at position 2”.
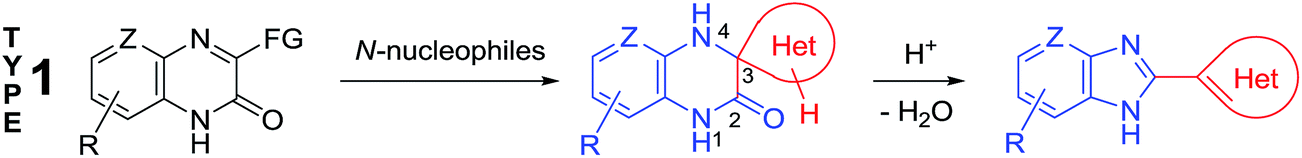 | ||
| Scheme 75 Schematical presentation of the rearrangement TYPE 1. | ||
The second one can be schematically presented as TYPE 2 on the Scheme 76: “Any of the spiro-derivatives of 1,2,3,4-tetrahydroquinoxalin-3-one without any mobile hydrogen atom in their spiro-forming component are on their way to the benzimidazolone derivative with the spiro-forming component at position 1”.
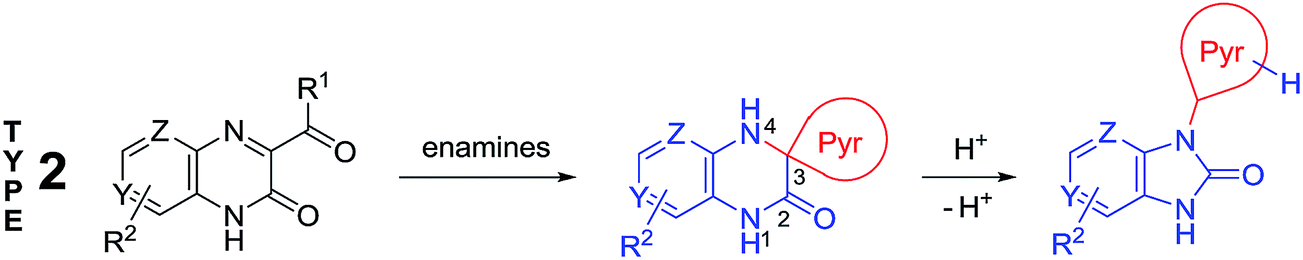 | ||
| Scheme 76 Schematical presentation of the rearrangement TYPE 2. | ||
Thus, the novel rearrangements presented raises no doubts as to its generality in the synthesis of benzimidazol(on)es. In accordance with the aim set both the first and the second component can be replaced when synthesizing any desired derivative of benzimidazole. Via these novel rearrangements 2-(benzimidazol-2-yl)-quinoxalines and their aza-analogues (2-(1H-imidazo[4,5-b]pyridin-2-yl)-3-arylquinoxalines), 2,3-bis(benzimidazol-2-yl)quinoxalines, 2-(pyrazin-2-yl)benzimidazoles, 2-(imidazol-4-yl)benzimidazoles, 2-(3-arylindolizin-2-yl)benzimidazole, 2-(pyrazol-3-yl)benzimidazoles, 2-(pyrrol-3-yl)benzimidazoles, 2-(benzimidazol-2-yl)quinolines, 4-(benzimidazol-2-yl)quinolines and their condensed analogues (benzimidazolo[2,1-a]pyrrolo[3,4-c]quinoline), N-pyrrolylbenzimidazol-2(3H)-ones and their aza analogues (N-pyrrolyl-1H-imidazo[5,4-b]pyridin-2(3H)-ones, N-pyrrolyl-1H-imidazo[4,5-c]pyridin-2(3H)-ones), N-(1H-indazol-3-yl)benzimidazol-2(3H)-ones have been synthesized to date. These derivatives are shown in the review. We hope that in future these new rearrangements, based on the rearrangement of quinoxalin(on)e derivatives into benzimidazol(on)e derivatives, will find their worthy place in the chemistry of heterocyclic compounds, and will serve as a simple and efficient method for the synthesis of various benzimidazole derivatives, along with the classical Phillips–Ladenburg and Weidenhagen reactions.
Acknowledgements
I thank the Russian Foundation for Basic Research (grant no. 13-03-00123-a) for financial support of our work. For help in the preparation of this article I thank Nataliya A. Zhukova and the authors of the dissertations D. F. Saifina (ref. 89), A. A. Kalinin (ref. 90), O. G. Isaykina (ref. 91), A. M. Murtazina (ref. 111) and V. R. Galimullina (ref. 121) and all my co-workers whose names appeared in the references.Notes and references
- (a) F. Hobrecker, Ber. Dtsch. Chem. Ges., 1872, 5, 290 Search PubMed; (b) J. Wright, Chem. Rev., 1951, 48, 397 CrossRef CAS PubMed; (c) K. Hofmann, The Chemistry of Heterocyclic Compounds, Imidazole and its Derivatives, Part 1, Interscience, London, 1953 Search PubMed.
- For selected reviews, see: (a) M. R. Grimmet, in Comprehensive Heterocyclic Chemistry, ed. K. T. Potts, Pergamon, Oxford, 1984, vol. 4 Search PubMed; (b) M. R. Grimmet, in Comprehensive Heterocyclic Chemistry II, ed. I. Shinkai, Pergamon, Oxford, 1996, vol. 3 Search PubMed.
- For selected reviews, see: (a) N. Preston, Chem. Rev., 1974, 74, 279 CrossRef; (b) R. J. Sundberg and R. B. Martin, Chem. Rev., 1974, 74, 471 CrossRef CAS.
- J. A. Asensio, E. M. Sanchez and P. Gomez-Romero, Chem. Soc. Rev., 2010, 39, 3210 RSC.
- Ionic Liquids in Synthesis, ed. P. Wasserscheid and T. Welton, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed.
- S. Hirashima, T. Suzuki, S. Noji, I. Ando, M. Komatsu, S. Ikede and H. Hashimoto, J. Med. Chem., 2006, 49, 4721 CrossRef CAS PubMed.
- U. Scheffer, A. Strick, V. Ludwig, S. Peter, E. Kalden and M. W. Gobel, J. Am. Chem. Soc., 2005, 127, 2211 CrossRef CAS PubMed.
- E. Sener, I. Yalcin, O. Temiz, I. Oren, A. Akin and N. Ucarturk, Il Farmaco, 1997, 52, 99 CAS.
- P. Sharma, A. Kumar and M. Sharma, Eur. J. Med. Chem., 2006, 41, 833 CrossRef CAS PubMed.
- P. Lindberg, P. Nordberg, T. Alminger, A. Brandstrom and B. Wallmark, J. Med. Chem., 1986, 29, 1327 CrossRef CAS PubMed.
- R. Mannhold, Drugs Future, 1985, 10, 570 CrossRef.
- (a) N. Singh, A. Pandurangan, K. Rana, P. Anand, A. Ahmad and A. K. Tiwari, Int. Curr. Pharm. J., 2012, 1, 119 CAS; (b) C. Mukhopadhyay, S. Ghosh, S. Sengupta and S. De, RSC Adv., 2011, 1, 1033 RSC; (c) S. Demirayak, I. Kayagil and L. Yurttas, Eur. J. Med. Chem., 2011, 46, 411 CrossRef CAS PubMed; (d) J. E. Payne, C. Bonnefous, K. T. Symons, P. M. Nguyen, M. Sablad, N. Rozenkrants, Y. Zhang, L. Wang, N. Yazdani, A. K Shiau., S. A. Noble, P. Rix, T. S. Rao, C. A. Hassig and N. D. Smith, J. Med. Chem., 2010, 53, 7739 CrossRef CAS PubMed; (e) R. Sevak, A. Paul, S. Goswami and D. Santani, Pharmacol. Res., 2002, 46, 351 CrossRef CAS PubMed; (f) L. K. Labanauskas, A. B. Brukstus, P. G. Gaidelis, V. A. Bucinskaite, E. B. Udrenaite and V. K. Dauksas, Pharm. Chem. J., 2000, 34, 353 CrossRef CAS; (g) B. Can-Eke, M. O. Puskullu, E. Buyukbingol and M. Iscan, Chem.–Biol. Interact., 1998, 113, 65 CrossRef CAS PubMed.
- (a) S. Bhattacharya and P. Chaudhuri, Curr. Med. Chem., 2008, 15, 1762 CrossRef CAS PubMed; (b) M. Boiani and M. Gonz'alez, Mini-Rev. Med. Chem., 2005, 5, 409 CrossRef CAS PubMed; (c) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893 CrossRef CAS PubMed.
- H. S. A. Elzahabi, Eur. J. Med. Chem., 2011, 46, 4025 CrossRef CAS PubMed.
- L. J. Scott, C. J. Dunn, G. Mallarkey and M. Sharpe, Drugs, 2002, 62, 1503 CrossRef CAS PubMed.
- P. J. Venkatesan, J. Antimicrob. Chemother., 1998, 41, 145 CrossRef CAS PubMed.
- H. J. Al Muhaimeed, J. Int. Med. Res., 1997, 25, 175 CrossRef CAS PubMed.
- (a) S. Nakamura, Chem. Pharm. Bull., 1955, 3, 379 CrossRef CAS; (b) A. Zeynep, A. Mehmet, K. Canan, Y. Sulhiye, B. Erdem and G. Hakan, Arch. Pharm. Chem. Life Sci., 2006, 339, 74 CrossRef PubMed; (c) T. Meral, G. Hakan, E. Rahmiye, E. Recep, K. Engin and A. Nurten, Arch. Pharm. Pharm. Med. Chem., 1997, 330, 372 CrossRef; (d) Z. M. Nofal, H. H. Fahmy and H. S. Mohamed, Arch. Pharmacal Res., 2002, 25, 28 CrossRef CAS.
- A. C. Cuckler and K. C. Mezey, Arzneim. Forsch., 1966, 16, 411 CAS.
- (a) A. C. Hollinshead and P. K. Smith, J. Pharmacol. Exp. Ther., 1958, 123, 54 CAS; (b) D. C. O'Sullivan and A. K. Wallis, J. Med. Chem., 1972, 15, 103 CrossRef; (c) B. Simone, K. Mariola, G. Agata, K. Zygmunt, E. Henning, L. Paolo, G. Gilles and S. Frank, ARKIVOC, 2009, iii, 225 Search PubMed; (d) R. Zou, K. Ayres, J. Drach and L. Townsend, J. Med. Chem., 1996, 39, 3477 CrossRef CAS PubMed.
- (a) W. W. Kilgore and E. R. White, Bull. Environ. Contam. Toxicol., 1970, 5, 67 CrossRef CAS PubMed; (b) B. Maxwell and G. Brody, Appl. Microbiol., 1971, 21, 944 Search PubMed; (c) E. I. Elnima, M. U. Zubair and A. A. Al-Badr, Antimicrob. Agents Chemother., 1981, 19, 29 CrossRef CAS PubMed; (d) K. A. M. Walker, A. C. Braemer, S. Hitt, R. E. Jones and T. R. Mathews, J. Med. Chem., 1978, 21, 840 CrossRef CAS PubMed; (e) C. J. Going and V. W. Mayer, Mutat. Res., 1995, 343, 185 CrossRef.
- The Pesticide Manual, ed. C. Tomlin, Royal Society of Chemistry, British Crop Protection, 1994 Search PubMed.
- G. Kamlesh, B. Jamie, P. Austin, B. Julie, W. Leslie and P. Dulal, Biochemistry, 2004, 43, 6645 CrossRef PubMed.
- (a) O. Seckin, A. Dilek, Y. Sulhiye and G. Hakan, Bioorg. Med. Chem., 2005, 13, 1587 CrossRef PubMed; (b) G. Hakan, M. Tuncbilek, G. Ayhan and N. Altanlar, Il Farmaco, 1998, 53, 415 CrossRef; (c) T. Hisano, M. Ichikawa, K. Tsumoto and M. Tasaki, Chem. Pharm. Bull., 1982, 30, 2996 CrossRef CAS.
- M. Tuncbilek, T. Kiper and N. Altanlar, Eur. J. Med. Chem., 2009, 44, 1024 CrossRef CAS PubMed.
- (a) E. A. Ibrahim, A. M. E. Omar and M. A. Khalil, J. Pharm. Sci., 1980, 69, 1348 CrossRef CAS PubMed; (b) A. K. Piskin, A.-A. Zeynep, F. B. Atac, Y. Musdal and E. Buyukbingol, Turk. J. Biochem., 2009, 34, 39 CAS; (c) T. A. Heba, A. F. R. Fatma, M. R. Mostafa and I. E. Hoda, Eur. J. Med. Chem., 2010, 45, 2336 CrossRef PubMed; (d) S. Mohammad, M. M. Abdullah, M. A. Bakht and M. Jaseela, Eur. J. Med. Chem., 2010, 45, 114 CrossRef PubMed; (e) N. Boufatah, A. Gellis, J. Maldonado and P. Vanelle, Tetrahedron, 2004, 60, 9131 CrossRef CAS.
- S. Alper, O. Temiz, E. Sener and I. Yalcin, Il Farmaco, 2003, 58, 497 CrossRef CAS PubMed.
- K.-K. Wong, Recent Pat. Anti-Cancer Drug Discovery, 2009, 4, 28 CrossRef CAS PubMed.
- N. K. Haass, K. Sproesser, T. K. Nguyen, R. Contractor, A. Medina, K. L. Nathanson, M. Herlyn and K. S. M. Smalley, Clin. Cancer Res., 2008, 14, 230 CrossRef CAS PubMed.
- W. A. Denny, G. W. Rewcastle and B. C. Baguley, J. Med. Chem., 1990, 33, 814 CrossRef CAS PubMed.
- (a) Z. Ates, B. C. Eke, S. Suzen, E. Buyukbingol and M. Iscan, Il Farmaco, 1997, 52, 703 CAS; (b) Z. Ates-Alagoz and E. Buyukbingol, Heterocycl. Commun., 2001, 7, 455 CrossRef CAS; (c) Z. Ates-Alagoz, B. C. Eke, T. Coban, M. Iscan and E. Buyukbingol, Arch. Pharm., 2004, 337, 188 CrossRef CAS PubMed.
- (a) S. Budow, M. Kozlowska, A. Gorska, Z. Kazimierczuk, H. Eickmeier, P. La Colla, G. Gosselin and F. Seela, ARKIVOC, 2009, iii, 225 Search PubMed; (b) M. T. Migawa, J. Girardet, J. A. Walker, G. W. Koszalka, S. D. Chamberlain, J. C. Drach and L. B. Townsend, J. Med. Chem., 1998, 41, 1242 CrossRef CAS PubMed; (c) A. R. Porcari, R. V. Devivar, L. S. Kucera, J. C. Drach and L. B. Townsend, J. Med. Chem., 1998, 41, 1252 CrossRef CAS PubMed.
- M. Andrzejewska, L. Yepez-Mulia, R. Cedillo-Rivera, A. Tapia, L. Vilpo, J. Vilpo and Z. Kazimierczuk, Eur. J. Med. Chem., 2002, 37, 973 CrossRef CAS PubMed.
- A. Pinar, P. Yurdakul, I. Yildiz, O. Temiz-Arpaci, N. L. Acan, E. Aki-Sene and I. Yalcin, Biochem. Biophys. Res. Commun., 2004, 317, 670 CrossRef CAS PubMed.
- H. M. Refaat, Eur. J. Med. Chem., 2010, 45, 2949 CrossRef CAS PubMed.
- (a) W. Wienen, N. Hauel, J. C. A. Van Meel, B. Narr, U. Ries and M. B. Entzeroth, J. Pharmacol., 1993, 110, 245 CAS; (b) A. J. Battershill and L. J. Scott, Drugs, 2006, 66, 51 CrossRef CAS PubMed; (c) K. J. McClellan and A. Markham, Drugs, 1998, 56, 1039 CrossRef CAS PubMed.
- (a) R. Cernes, M. Mashavi and R. Zimlichman, Vasc. Health Risk Manage., 2011, 7, 749 CAS; (b) M. Burnier and H. R. Brunner, Lancet, 2000, 355, 637 CrossRef CAS PubMed.
- (a) S. C. Benson, H. A. Pershadsingh, C. I. Ho, A. Chittiboyina, P. Desai, M. Pravenec, N. Qi, J. Wang, M. A. Avery and T. W. Kurtz, Hypertension, 2004, 43, 993 CrossRef CAS PubMed; (b) R. A. Benndorf, T. Rudolph, D. Appel, E. Schwedhelm, R. Maas, F. Schulze, E. Silberhorn and R. H. Boger, Metab., Clin. Exp., 2006, 55, 1159 CrossRef CAS PubMed; (c) J. F. E. Mann, R. E. Schmieder, M. McQueen, L. Dyal, H. Schumacher, J. Pogue, X. Wang, A. Maggioni, A. Budaj, S. Chaithiraphan, K. Dickstein, M. Keltai, K. Metsärinne, A. Oto, A. Parkhomenko, L. S. Piegas, T. L. Svendsen, K. K. Teo and S. Yusuf, Lancet, 2008, 372, 547 CrossRef CAS PubMed.
- (a) A. Carta, S. Piras, G. Loriga and G. Paglietti, Mini-Rev. Med. Chem., 2006, 6, 1179 CrossRef CAS PubMed; (b) B. P. Zambrowicz and A. T. Sands, Nat. Rev. Drug Discovery, 2003, 2, 38 CrossRef CAS PubMed.
- S. Ohta, Y. Naita, T. Yuasa, S. Hatakeyama, M. Kobayashi, K. Kaibe, I. Kawasaki and M. Yamashita, Chem. Pharm. Bull., 1991, 39, 2787 CrossRef CAS.
- Y. Kuo-Long, R. L. Civiello, K. D. Combrink, H. B. Gulgeze, N. Sin, X. Wang, A. Nicholas and B. L. Venables, WO Pat., 2001/95910 A1, 2001.
- D. F. Smith, L. Whitesell and E. Katsanis, Pharmacol. Rev., 1998, 50, 493 CAS.
- Lekarstvennye Sredstva, ed. M. D. Mashkovskiy, Novaya Volna, Moscow, 2008 Search PubMed.
- M. Negwer and H.-G. Sharnow, Organic-Chemical Drugs and Their Synonyms, Wiley-VCH, Weinheim, 2001 Search PubMed.
- J. A. Joule and K. Mills, Heterocyclic Chemistry, Wiley, 2010, pp. 507–508 Search PubMed.
- (a) D. N. Gray, J. Heterocycl. Chem., 1970, 7, 947 CrossRef CAS; (b) R. L. Hudkins, Heterocycles, 1995, 41, 1045 CrossRef CAS.
- (a) V. Balasubramaniyan, P. Balasubramaniyan and S. V. Patil, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1990, 29, 124 Search PubMed; (b) M. S. Salakhov, V. S. Umaeva, Y. S. Salakhova and S. S. Idrisova, Zh. Org. Khim., 1999, 35, 421 Search PubMed; (c) M. R. Grimmett, in Best Synthetic Methods, ed. O. Meth-Cohn and A. R. Katritzky, Academic Press, New York, 1997 Search PubMed.
- R. A. Ogg and F. W. Bergstrom, J. Am. Chem. Soc., 1931, 53, 1846 CrossRef CAS.
- E. C. Taylor and A. McKillop, J. Org. Chem., 1965, 30, 2858 CrossRef CAS.
- E. S. Lane, J. Chem. Soc., 1955, 1079 RSC.
- (a) M. J. Haddadin and C. H. Issidorides, Tetrahedron Lett., 1967, 8, 753 CrossRef; (b) A. A. Jarrer, S. S. Halawi and M. J. Haddadin, Heterocycles, 1976, 4, 1077 CrossRef.
- (a) F. Krohnke, Justus Liebigs Ann. Chem., 1957, 604, 203 CrossRef CAS; (b) J. Streith and C. Sigwatt, Tetrahedron Lett., 1966, 13, 1347 CrossRef; (c) J. S. Splitter and M. Calvin, J. Org. Chem., 1965, 30, 3427 CrossRef CAS.
- Y. Ahmad, M. S. Habib, A. Mohammady, B. Bakhtiari and S. A. Shamsi, J. Org. Chem., 1968, 33, 201 CrossRef CAS.
- P. T. Lont and H. C. Van der Plas, Recl. Trav. Chim. Pays-Bas, 1972, 91, 850 CrossRef CAS.
- G. W. H. Cheeseman and M. Rafig, J. Chem. Soc. C, 1971, 452 RSC.
- A. Albert and J. J. McCormack, J. Chem. Soc., 1965, 6930 RSC.
- R. A. Burrell, J. M. Cox and E. G. Savins, J. Chem. Soc., Perkin Trans. 1, 1973, 2707 RSC.
- R. A. Abramovitch and B. W. Cue, Heterocycles, 1973, 1, 227 CrossRef CAS.
- E. Hayashi and C. Iijima, Yakugaki Zasshi, 1962, 82(8), 1093 ( Chem. Abstr. , 1963 , 58 , 4551 ) CAS.
- Y. Ahmad, M. Habib, S. Ziauddin and B. Bakhtiari, J. Org. Chem., 1966, 31, 2613 CrossRef CAS PubMed.
- E. Hayashi and Y. Miura, Yakudaku Zasshi, 1967, 87, 648 ( Chem Abstr. , 1967 , 67 , 90775s ) CAS.
- (a) A. A. Kalinin, V. A. Mamedov and Y. A. Levin, Chem. Heterocycl. Compd., 2000, 36, 882 CrossRef CAS; (b) V. A. Mamedov, A. A. Kalinin, A. T. Gubaidullin, A. V. Chernova, I. A. Litvinov, A. Ya and R. R. Shagidullin, Russ. Chem. Bull., 2004, 53, 164 CrossRef CAS; (c) V. A. Mamedov, A. A. Kalinin, A. T. Gubaidullin, E. A. Gorbunova and I. A. Litvinov, Russ. J. Org. Chem., 2006, 42, 1532 CrossRef CAS; (d) A. A. Kalinin, O. G. Isaykina and V. A. Mamedov, Chem. Heterocycl. Compd., 2007, 43, 1307 CrossRef CAS; (e) V. A. Mamedov, D. F. Saifina, I. K. Rizvanov and A. T. Gubaidullin, Tetrahedron Lett., 2008, 49, 4644 CrossRef CAS; (f) V. A. Mamedov, N. A. Zhukova, T. N. Beschastnova, A. T. Gubaidullin, A. A. Balandina and S. K. Latypov, Tetrahedron, 2010, 66, 9745 CrossRef CAS.
- (a) V. A. Mamedov, A. A. Kalinin, A. T. Gubaidullin, I. A. Litvinov and Y. A. Levin, Chem. Heterocycl. Compd., 2002, 38, 1504 CrossRef CAS; (b) E. A. Gorbunova and V. A. Mamedov, Russ. J. Org. Chem., 2006, 42, 1528 CrossRef CAS; (c) V. A. Mamedov, A. M. Murtazina, D. I. Adgamova, N. A. Zhukova, T. N. Beschastnova, S. V. Kharlamov, I. K. Rizvanov and S. K. Latypov, J. Heterocycl. Chem., 2014, 51, 1664 CrossRef CAS.
- V. A. Mamedov, A. A. Kalinin, A. T. Gubaidullin, O. G. Isaikina and I. A. Litvinov, Russ. J. Org. Chem., 2005, 41, 599 CrossRef CAS.
- O. Hinsberg, Ber. Dtsch. Chem. Ges., 1884, 17, 318 CrossRef.
- S. von Niementowski, Ber. Dtsch. Chem. Ges., 1897, 30, 3062 CrossRef CAS.
- E. L. Hölljes and E. C. Wagner, J. Org. Chem., 1944, 9, 31 CrossRef.
- F. E. King and R. M. Acheson, J. Chem. Soc., 1949, 1396 RSC.
- (a) C. T. Brain and S. A. Brunton, Tetrahedron Lett., 2002, 43, 1893 CrossRef CAS; (b) C. T. Brain and J. T. Steer, J. Org. Chem., 2003, 68, 6814 CrossRef CAS PubMed.
- (a) J. B. Wright, Chem. Rev., 1951, 48, 397 CrossRef CAS PubMed; (b) R. C. Elderfield and F. J. Kreysa, J. Am. Chem. Soc., 1948, 70, 44 CrossRef CAS.
- R. Weidenhagen, Ber. Dtsch. Chem. Ges., 1936, 69, 2263 CrossRef.
- P. L. Beaulieu, B. Haché and E. von Moos, Synthesis, 2003, 1683 CrossRef CAS.
- J. I. G. Cadogan, R. Marshall, D. M. Smith and M. J. Todd, J. Chem. Soc. C, 1970, 2441 RSC.
- S. Tolari, S. Cenini, C. Crotti and E. Gianella, J. Mol. Catal., 1994, 87, 203 CrossRef.
- (a) W. Dohle, A. Staubitz and P. Knochel, Chem.–Eur. J., 2003, 9, 5323 CrossRef CAS PubMed; (b) R. J. Sundberg, J. Org. Chem., 1965, 30, 3604 CrossRef CAS; (c) R. J. Sundberg and T. Yamazaki, J. Org. Chem., 1967, 32, 290 CrossRef CAS.
- (a) V. A. Mamedov, A. M. Murtazina, A. T. Gubaidullin, E. A. Khafizova, I. K. Rizvanov and I. A. Litvinov, Russ. Chem. Bull., Int. Ed., 2010, 59, 1645 CrossRef CAS; (b) V. A. Mamedov, E. A. Khafizova, A. T. Gubaidullin, A. M. Murtazina, D. I. Adgamova, A. I. Samigullina and I. A. Litvinov, Russ. Chem. Bull., Int. Ed., 2011, 60, 368 CrossRef CAS.
- G. Sarodnick and G. Kempter, Zeitschrift für Chemie, 1982, 22, 300 CrossRef CAS.
- (a) T. Mizutani, K. Yoshida and S. Kawazoe, Drug Metab. Dispos., 1994, 22, 750 CAS; (b) E. Novellino, B. Cosimelli, M. Ehlardo, G. Greco, M. Iadanza, A. Lavecchia, M. G. Rimoli, A. Sala, A. Da Settimo, G. Primofiore, F. Da Settimo, S. Taliani, C. La Motta, K. N. Klotz, D. Tuscano, M. L. Trincavelli and C. Martini, J. Med. Chem., 2005, 48, 8253 CrossRef CAS PubMed.
- E. Lippmann and W. Shilov, Collect. Czech. Chem. Commun., 1984, 49, 1304 CrossRef CAS.
- M. J. Climent, A. Corma, S. Iborra and S. Martínez-Silvestre, ChemCatChem, 2013, 5, 3866 CrossRef CAS.
- (a) A. Domling, Chem. Rev., 2006, 106, 17 CrossRef PubMed; (b) C. Hulme and J. Dietrich, Mol. Diversity, 2009, 13, 195 CrossRef CAS PubMed; (c) L. El Kaim and L. Grimaud, Tetrahedron, 2009, 65, 2153 CrossRef CAS; (d) L. Banfi, R. Riva and A. Basso, Synlett, 2010, 1, 23 CrossRef; (e) C. Hulme and V. Gore, Curr. Med. Chem., 2003, 44, 1947 Search PubMed; (f) Z. Xu, M. Ayaz, A. Capelli and C. Hulme, ACS Comb. Sci., 2012, 14, 460 CrossRef CAS PubMed; (g) Z. Xu, F. De Moliner, A. P. Capelli and C. Hulme, Angew. Chem., Int. Ed., 2012, 51, 8037 CrossRef CAS PubMed.
- (a) C. Hulme, J. Peng, S. Y. Tang, C. J. Burns, I. Morize and R. Labaudiniere, J. Org. Chem., 1998, 63, 8021 CrossRef CAS; (b) Y. Huang, K. Khoury, T. Chanas and A. Doemling, Org. Lett., 2012, 14, 5916 CrossRef CAS PubMed.
- (a) C. Hulme, J. Peng, B. Louridas, P. Menard, P. Krolikowski and N. V. Kumar, Tetrahedron Lett., 1998, 39, 8047 CrossRef CAS; (b) C. Hulme, J. Peng, G. Morton, J. M.Salvino, T. Herpin and R. Labaudiniere, Tetrahedron Lett., 1998, 39, 7227 CrossRef CAS.
- C. Hulme, L. Ma, J. Romano and M. Morrissette, Tetrahedron Lett., 1999, 40, 7925 CrossRef CAS.
- C. Hulme, L. Ma, M. P. Cherrier, J. J. Romano, G. Morton, C. Duquenne, J. Salvino and R. Labaudiniere, Tetrahedron Lett., 2000, 41, 1883 CrossRef CAS.
- (a) C. Hulme, L. Ma, J. J. Romano, G. Morton, S.-Y. Tang, M.-P. Cherrier, S. Choi, J. Salvino and R. Labaudiniere, Tetrahedron Lett., 2000, 41, 1889 CrossRef CAS; (b) S. Pakornwit and M. Krasivan, Tetrahedron Lett., 2014, 55, 2299 CrossRef.
- M. Ayaz, Z. Xu and C. Hulme, Tetrahedron Lett., 2014, 55, 3406 CrossRef CAS PubMed.
- S. Gunawan, M. Ayaz, F. De Moliner, B. Frett, C. Kaiser, N. Patrick, Z. Xu and C. Hulme, Tetrahedron, 2012, 68, 5606 CrossRef CAS PubMed.
- D. F. Saifina, PhD dissertation, A. E. Arbuzov Institute of Organic and Physical Chemistry of Kazan Research Center of the Russian Academy of Sciences, Kazan, Russia, 2009.
- A. A. Kalinin, PhD dissertation, Kazan State University, Kazan, Russia, 2000.
- O. G. Isaykina, PhD dissertation, A. E. Arbuzov Institute of Organic and Physical Chemistry of Kazan Research Center of the Russian Academy of Sciences, Kazan, Russia, 2007.
- (a) P. N. Preston, Chem. Rev., 1974, 74, 279 CrossRef CAS; (b) A. Ladenburg, Chem. Ber., 1875, 8, 677 CrossRef; (c) M. A. Phillips, J. Chem. Soc., 1928, 13, 172 RSC; (d) M. A. Phillips, J. Chem. Soc., 1928, 13, 2393 RSC; (e) A. W. White, R. Almassy, A. H. Calvert, N. J. Curtin, R. J. Griffin, Z. Hostomsky, K. Maegley, D. R. Newell and B. T. Golding, J. Med. Chem., 2000, 43, 4084 CrossRef CAS PubMed; (f) I. M. Elchaninov and M. M. Elchaninov, Russ. J. Org. Chem., 2014, 50, 1145 CrossRef CAS; (g) B. H. Kim, R. Han, T. H. Han, Y. M. Jun, W. Baik and B. M. Lee, Heterocycles, 2002, 57, 5 CrossRef CAS.
- J. A. Joule and K. Mills, Heterocyclic Chemistry, Wiley, 2010, pp. 462–463 Search PubMed.
- S. Kanoktanaporn, J. A. H. MacBride and T. J. King, J. Chem. Res., 1980, 406, 4901 Search PubMed.
- Y. Kurasawa, S. Shimabukuro, Y. Okamoto and A. Takada, J. Heterocycl. Chem., 1985, 22, 1461 CrossRef CAS.
- A. Hassner and I. Namboothiri, Organic Syntheses Based on Name Reactions, Elsevier, Amsterdam, 2012, pp. 299–300 Search PubMed.
- (a) Y. Kurasawa and A. Takada, Heterocycles, 1980, 14, 281 CrossRef CAS; (b) Y. Kurasawa, Y. Okamoto, K. Ogura and A. Takada, J. Heterocycl. Chem., 1985, 22, 661 CrossRef CAS; (c) Y. Kurasawa, J. Satoh, M. Ogura, Y. Okamoto and A. Takada, Heterocycles, 1984, 22, 1531 CrossRef CAS.
- L. J. Bellamy, The Infra-red Spectra of Complex Molecules, Wiley, New York, 1975 Search PubMed.
- V. A. Mamedov, A. M. Murtazina, N. A. Zhukova, T. N. Beschastnova, I. K. Rizvanov and S. K. Latypov, Tetrahedron, 2014, 70, 7567 CrossRef CAS.
- (a) N. Kornblum, W. J. Jones and G. J. Anderson, J. Am. Chem. Soc., 1959, 81, 4113 CrossRef CAS; (b) N. Kornblum and H. W. Frazier, J. Am. Chem. Soc., 1966, 88, 865 CrossRef CAS; (c) S. Chandrasekhar and M. Sridhar, Tetrahedron Lett., 2000, 41, 5423 CrossRef CAS; (d) B. K. Bettadaiah, K. N. Burudutt and P. Srinivas, J. Org. Chem., 2003, 68, 2460 CrossRef CAS PubMed.
- March's Advanced Organic Chemistry, ed. M. B. Smith, Hooboken, New Jersey, 2013, pp. 1481–1483 Search PubMed.
- V. A. Mamedov, N. A. Zhukova, V. V. Sykaev, A. T. Gubaidullin, T. N. Beschastnova, D. I. Adgamova, A. I. Samigullina and S. K. Latypov, Tetrahedron, 2013, 69, 1403 CrossRef CAS.
- V. A. Mamedov, N. A. Zhukova, T. N. Beschastnova, E. I. Zakirova, S. F. Kadyrova, E. V. Mironova, A. G. Nikonova, S. K. Latypov and I. A. Litvinov, Tetrahedron Lett., 2012, 53, 292 CrossRef CAS.
- (a) O. Hinsberg, Liebigs Ann. Chem., 1887, 237, 368 CrossRef; (b) S. N. Murthy, B. Madhav and Y. V. D. Nageswar, Helv. Chim. Acta, 2010, 93, 1216 CrossRef CAS.
- G. W. H. Cheeseman and R. F. Cookson, in Condensed Pyrazines, ed. A. Weissberger and E. C. Taylor, Wiley-Interscience, New York, 1979 Search PubMed.
- (a) Y. Kubota, T. Shibata, E. Babamoto-Horiguchi, J. Uehara, K. Funabiki, S. Matsumoto, M. Ebihara and M. Matsui, Tetrahedron, 2009, 65, 2506 CrossRef CAS; (b) B. Gao, Q. Zhou, Y. Geng, Y. Cheng, D. Ma, Z. Xie, L. Wang and F. Wang, Mater. Chem. Phys., 2006, 99, 247 CrossRef CAS; (c) R. Faust and C. Weber, Tetrahedron, 1997, 53, 14655 CrossRef CAS.
- (a) V. A. Mamedov, D. F. Saifina, A. T. Gubaidullin, A. F. Saifina and I. K. Rizvanov, Tetrahedron Lett., 2008, 49, 6231 CrossRef CAS; (b) V. A. Mamedov, A. M. Murtazina, A. T. Gubaidullin, E. A. Hafizova and I. K. Rizvanov, Tetrahedron Lett., 2009, 50, 5186 CrossRef CAS; (c) V. A. Mamedov, D. F. Saifina, A. T. Gubaidullin, V. R. Ganieva, S. F. Kadyrova, D. V. Rakov, I. K. Rizvanov and O. G. Sinyashin, Tetrahedron Lett., 2010, 51, 6503 CrossRef CAS.
- (a) V. A. Mamedov, N. A. Zhukova, T. N. Beschastnova, A. T. Gubaidullin, D. V. Rakov and I. K. Rizvanov, Tetrahedron Lett., 2011, 52, 4280 CrossRef CAS; (b) V. A. Mamedov, N. A. Zhukova, T. N. Beschastnova and A. T. Gubaidullin, Russ. Chem. Bull., Int. Ed., 2011, 60, 933 CrossRef CAS.
- V. A. Mamedov, D. F. Saifina, A. T. Gubaidullin, A. F. Saifina, I. K. Rizvanov and V. R. Ganieva, Russ. Chem. Bull., Int. Ed., 2009, 58, 1986 CrossRef CAS.
- A. M. Murtazina, PhD dissertation, A. E. Arbuzov Institute of Organic and Physical Chemistry of Kazan Research Center of the Russian Academy of Sciences, Kazan, Russia, 2010.
- (a) M. Behforouz, J. L. Bolan and M. S. Flynt, J. Org. Chem., 1985, 50, 1186 CrossRef CAS; (b) J. Buckingham, Q. Rev., Chem. Soc., 1969, 23, 37 RSC; (c) L. A. Audrieth and B. A. Ogg, The Chemistry of Hydrazines, Wiley, New York, 1951 Search PubMed.
- (a) M. B. Smith, March's Advanced Organic Chemistry, Hooboken, New Jersey, 2013, pp. 1067–1248 Search PubMed; (b) C. D. Gutsche, The Chemistry of Carbonyl Compounds, Prentice Hall, Englewood Cliffs, NJ, 1967 Search PubMed.
- (a) L. Knorr, Ber. Dtsch. Chem. Ges., 1884, 17, 1635 CrossRef; (b) A. Castro, D. D. Giannin and W. F. Greenlee, J. Org. Chem., 1970, 35, 2815 CrossRef CAS; (c) A. F. Mironov, K. K. Alarkon and R. P. Evstigneeva, Chem. Heterocycl. Compd., 1973, 1487 CrossRef.
- (a) P. T. Parvatkar, P. S. Parameswaran and S. G. Tilve, J. Org. Chem., 2009, 74, 8369 CrossRef CAS PubMed; (b) L. Zhu and M. Zhang, J. Org. Chem., 2004, 69, 7371 CrossRef CAS PubMed; (c) G. E. Ham, J. Org. Chem., 1964, 29, 3052 CrossRef CAS; (d) E. Byun, B. Hong, K. A. De Castro, M. Lim and H. Rhee, J. Org. Chem., 2007, 72, 9815 CrossRef CAS PubMed; (e) J. Marco-Contelles, E. Pérez-Mayoral, A. Samadi, M. do Carmo Carreiras and E. Soriano, Chem. Rev., 2009, 109, 2652 CrossRef CAS PubMed; (f) M. Bian, W. Yao, H. Ding and C. Ma, J. Org. Chem., 2010, 75, 269 CrossRef CAS PubMed; (g) T. Newhouse, C. A. Lewis, K. J. Eastman and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 7119 CrossRef CAS PubMed.
- (a) E. Bandini, G. Corda, A. D'Aurizio and M. Panunzio, Tetrahedron Lett., 2010, 51, 933 CrossRef CAS; (b) V. Gouverneur and L. Ghosez, Tetrahedron, 1996, 52, 7585 CrossRef CAS; (c) Z. Bouaziz, P. Nebois and H. Fillion, Tetrahedron, 1995, 51, 4057 CrossRef CAS; (d) C. R. Berry and R. P. Hsung, Tetrahedron, 2004, 60, 7629 CrossRef CAS; (e) F. Palacios and G. Rubiales, Tetrahedron Lett., 1996, 37, 6379 CrossRef CAS; (f) I. A. Motortna and D. S. Grierson, Tetrahedron Lett., 1999, 40, 7211 CrossRef; (g) M. A. Cuellar, L. K. Alegria, Y. A. Prieto, M. J. Cortes, R. A. Tapia and M. D. Preite, Tetrahedron Lett., 2002, 43, 2127 CrossRef CAS.
- V. A. Mamedov, A. A. Kalinin, V. V. Yanilkin, A. T. Gubaidullin, S. K. Latypov, A. A. Balandina, O. G. Isaikina, A. V. Toropchina, N. V. Nastapova, N. A. Iglamova and I. A. Litvinov, Russ. Chem. Bull., Int. Ed., 2005, 54, 2616 CrossRef CAS.
- V. A. Mamedov and N. A. Zhukova, in Progress in Heterocyclic Chemistry, ed. G. W. Gribble and J. A. Joule, Elsevier, Amsterdam, 2013, vol. 25, pp. 1–45 Search PubMed.
- V. A. Mamedov and A. M. Murtazina, Russ. Chem. Rev., 2011, 80, 397 CrossRef CAS.
- (a) V. A. Mamedov, V. R. Galimullina, N. A. Zhukova, S. F. Kadyrova, E. V. Mironova, I. K. Rizvanov and S. K. Latypov, Tetrahedron Lett., 2014, 55, 4319 CrossRef CAS; (b) V. A. Mamedov, S. F. Kadyrova, N. A. Zhukova, V. R. Galimullina, F. M. Polyancev and S. K. Latypov, Tetrahedron, 2014, 70, 5934 CrossRef CAS.
- V. R. Galimullina, PhD dissertation, A. E. Arbuzov Institute of Organic and Physical Chemistry of Kazan Research Center of the Russian Academy of Sciences, Kazan, Russia, 2015.
- M. B. Smith, March's Advanced Organic Chemistry, Wiley, New York, 2001, pp. 1185–1187 Search PubMed.
- Z. Wang, Comprehensive Organic Name Reactions and Reagents, Wiley, Hoboken, New Jersey, 2009, pp. 1137–1142 Search PubMed.
- (a) Z. Rappoport, The Chemistry of Enamines, Wiley, Chichester, UK, 1994 CrossRef; (b) P. W. Hickmott, Tetrahedron, 1984, 40, 2989 CrossRef CAS; (c) P. W. Hickmott, Tetrahedron, 1982, 38, 3363 CrossRef CAS; (d) P. W. Hickmott, Tetrahedron, 1982, 38, 1975 CrossRef CAS.
- I. Wiedermannová, J. Slouka, O. Humpa and K. Lemr, J. Heterocycl. Chem., 2003, 40, 357 CrossRef.
- A. Lučka, I. Frišová and J. Slouka, Magn. Reson. Chem., 2007, 45, 46 CrossRef PubMed.
- I. Frišová, Z. Trávníček, J. Slouka and P. Cankař, ARKIVOC, 2011, 127 Search PubMed.
- V. A. Mamedov, N. A. Zhukova, A. I. Zamaletdinova, T. N. Beschastnova, M. S. Kadyrova, I. K. Rizvanov, V. V. Syakaev and S. K. Latypov, J. Org. Chem., 2014, 79, 9161 CrossRef CAS PubMed.
- (a) S. Allu, S. Selvakumar and V. K. Singh, Tetrahedron Lett., 2010, 51, 446 CrossRef CAS; (b) O. Andrei, A. Alexakis and G. Bernardinelli, Org. Lett., 2003, 5, 2559 CrossRef PubMed; (c) S. Harada, N. Kumagai, T. Kinoshita, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2003, 9, 2582 CrossRef PubMed; (d) S. Boncel, M. Mączka and K. Z. Walczak, Tetrahedron, 2010, 43, 8450 CrossRef.
- V. A. Mamedov, N. A. Zhukova, T. N. Beschastnova, V. V. Syakaev, D. B. Krivolapov, E. V. Mironova, A. I. Zamaletdinova, I. K. Rizvanov and S. K. Latypov, J. Org. Chem., 2015, 80, 1375 CrossRef CAS PubMed.
- (a) Y. Naruse, T. Suzuki and S. Inagaki, Tetrahedron Lett., 2005, 46, 6937 CrossRef CAS; (b) G. Hilt and J. Treutwein, Angew. Chem., 2007, 46, 8500 CrossRef CAS PubMed; (c) R. Shen, S. Zhu and X. Huang, J. Org. Chem., 2009, 74, 4118 CrossRef CAS PubMed.
- (a) G. E. Keck and R. R. Webb, J. Am. Chem. Soc., 1981, 103, 3173 CrossRef CAS; (b) G. E. Keck and R. Webb, Tetrahedron Lett., 1979, 20, 1185 CrossRef; (c) W. Oppolzer, E. Pfenninger and K. Keller, Helv. Chim. Acta, 1973, 56, 1807 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |