DOI:
10.1039/C6RA02299E
(Paper)
RSC Adv., 2016,
6, 40664-40675
A novel heterojunction photocatalyst, Bi2SiO5/g-C3N4: synthesis, characterization, photocatalytic activity, and mechanism†
Received
26th January 2016
, Accepted 4th April 2016
First published on 7th April 2016
Abstract
A new type of heterojunction photocatalyst, Bi2SiO5/g-C3N4, was prepared using a controlled hydrothermal method. The structure and morphology of the Bi2SiO5/g-C3N4 photocatalyst were characterized by XRD, HR-TEM, FE-SEM-EDS, HR-XPS, FT-IR, PL, BET, EPR, and UV-Vis-DRS. The obtained Bi2SiO5/g-C3N4 photocatalyst exhibits enhanced photocatalytic activity on the decolorization of crystal violet (CV) under visible-light irradiation. In particular, the catalytic performance illustrates the best reaction rate constant of 0.1257 h−1 using Bi2SiO5/g-C3N4 as the photocatalyst, which is 5 and 3 times higher than the reaction rate constants of Bi2SiO5 and g-C3N4 as photocatalysts, respectively. This study shows that Bi2SiO5/g-C3N4 can be used to suppress the recombination of photoinduced electron–hole pairs and contribute to the enhanced photocatalytic efficiency of semiconductors in the visible light-driven catalysis. The quenching effects of different scavengers, and EPR results demonstrate that the reactive O2˙− plays the major role and ˙OH, h+ and 1O2 play minor roles in CV degradation. The probable photodegradation mechanisms are proposed and discussed.
1. Introduction
In the past decade, heterogeneous photocatalysis for environmental remediation and solar energy conversion has aroused extensive interest. For the practical applications of photocatalysis, an environmentally powerful and cheap photocatalyst is an important component.1 As such, triphenylmethane dyes have found applications as colorants in industry and as antimicrobial agents.2 However, great troubles have arisen with respect to the thyroid peroxidase-catalyzed oxidation of the triphenylmethane (triarylmethane) class of dyes because the reactions might produce various N-de-alkylated primary and secondary aromatic amines, with structures similar to aromatic amine carcinogens.3 The degradation of CV, a cationic triarylmethane dye, was studied using several systems that generated active species, including Bi2WO6,4 TiO2,5–10 ZnO,11–13 SrTiO3,14 BiOX/BiOY (X, Y = Cl, Br, I),15 BixAgyOz,16 BaTiO3,17 activated charcoal/Bi/ZnO,18 Ti(SO4)2,19 CdS/TiO2/rGO,20 CdS/ZnO,21 CuS/Bi2S3,22 bismuth silver oxide,23 Ag/AgVO3,24 n-BiVO4@p-MoS2,25 2D-Ag–TiO2,26 Au/TiO2/h-BN,27 NaYF4:Yb,Tm@TiO2/Ag (UC@TiO2/Ag),28 Ag@AgCl/ZnSn(OH)6,29 SiO2@TiO2,30 Zn3V2O8,31 Fe3O4@SiO2@TiO2@Ag,32 [(Me)2-2,2′-bipy]2Ag7I11,33 K2Ti8O17,34 In2S3,35 and Ag/Bi2O3/TiO2.36
Recently, Bi-based layered structure compounds, within the Aurivillius family, such as BiOX (X = Cl, Br, I),37,38 Bi2WO6,39 BiVO4,40 Bi4Ti3O12,41 etc., have been extensively researched as highly efficient photocatalysts due to their unique layered structures and high catalytic activities. It is thought that the Bi 6s and O 2p levels can make a largely dispersed hybridized valence band, which favors the mobility of photogenerated holes and oxidation reactions, inducing efficient separation of photogenerated electron–hole pairs and then improving photocatalytic efficiency.42
In the Bi2O3–SiO2 binary system, there are three compounds, Bi12SiO20, Bi4Si3O12 and Bi2SiO5,43–45 where Bi2SiO5 is one of the newly found compounds within the Aurivillius family. It has been described as a promising material with relatively good piezoelectric and dielectric properties and nonlinear optical effects. Recently, its luminescence properties were also reported. Pure Bi2SiO5 crystals were prepared by a novel method involving the melt-cooling process46 and the molten salt method in NaCl–Na2SO4 flux, using Bi2O3 and SiO2 as starting materials.47 Xie et al. also synthesized Bi4Si3O12 by a sol–gel method using Si(OC2H5)4 and Bi(NO3)3·5H2O as the precursors, and acetic acid as the solvent.48 He et al. prepared Bi12SiO20 by a chemical solution decomposition technique using Bi(NO3)3·5H2O and Si(OC2H5)4 as the starting materials for the degradation of Congo red.49 Duan's group synthesized Bi2SiO5 by hydrothermal methods, and it demonstrated excellent photocatalytic abilities and good stability during rhodamine B photodegradation under visible-light irradiation.50 Chen et al. showed the synthesis of Bi2SiO5 by a template-free hydrothermal method, and the Bi2SiO5 showed higher photocatalytic activity toward salicylic acid and benzene under UV-light irradiation.45 Police et al. reported porous Bi2SiO5 by an emulsion polymerization technique, for the degradation of isoproturon.51 Bi4Si3O12 nanofibers were also synthesized and tested for the degradation of methylene blue.52
Recently, the development of visible-light-driven photocatalysts has obtained considerable attention as an alternative in wastewater treatment. An effective and simple tactic to improve the photocatalytic activity of a photocatalyst is the incorporation of a heterostructure, because heterojunctions have great potential for tuning the desired electronic properties of photocatalysts and efficiently separating the photogenerated electron–hole pairs.48–50 So far, heterojunctions concerning Bi2SiO5, such as Bi4O5Br2/Bi24O31Br10/Bi2SiO5, have been reported, which exhibit enhanced photocatalytic efficiency.53 Therefore, it is feasible for BiOBr to be partly transformed into Bi2SiO5 via a thermodynamically favored route through the ion exchange reaction, which allows the exchange between the component ions and the incoming species,54 and consequently, the BiOBr/Bi2SiO5 heterojunction can be obtained. Wan et al. reported excellent photocatalytic activities of Bi2SiO5/AgI nanoplates for acid red G and formaldehyde.55 Liu et al. revealed that the photocatalytic activities of the Bi2S3/Bi2SiO5 heterojunctions were evaluated by degrading rhodamine B under visible light and UV-vis light irradiation.56 Lei et al. found that Ti-modified MoO3–Bi2SiO5/SiO2 exhibits excellent catalytic performance for the epoxidation of propylene by molecular oxygen.57
In the search for powerful and stable visible-light-driven photocatalysts, a polymeric semiconductor, graphitic carbon nitride (g-C3N4), has recently attracted great attention. The heptazine ring structure and high condensation degree enable the metal-free g-C3N4 to possess many advantages such as good physicochemical stability, as well as an appealing electronic structure combined with a medium-band gap (2.7 eV).58 These unique properties make g-C3N4 a promising candidate for visible light photocatalytic applications utilizing solar energy.
Graphitic carbon nitride is a well-known π-conjugated material for the improvement of the photogenerated electron–hole pair separation. More importantly, it is profoundly resistant to temperature and chemicals due to its s-triazine ring structure and high condensation. Besides, the g-C3N4 sheet is a soft polymer and can therefore be used as a coating material for other compounds. Recently, BiOI/g-C3N4,59 Ag3PO4@g-C3N4,60 and Zn2GeO4/g-C3N4 (ref. 61) composites have been synthesized in order to improve the photocatalytic activity of the materials. It is expected that functionalizing g-C3N4 nanosheets with bismuth silicates will not only combine the advantages of both bismuth silicate and g-C3N4 nanosheets, but will also result in new properties. However, no work on g-C3N4 based on bismuth silicate photocatalysts has been reported.
To the best of our knowledge, nanocomposite semiconductors consisting of Bi2SiO5 and g-C3N4 have not yet been reported in the literature. This is the first report that Bi2SiO5/g-C3N4 heterojunctions have been prepared by a template-free hydrothermal method and characterized by FE-SEM-EDS, XRD, HR-XPS, and UV-Vis-DRS. Through the degradation of CV in aqueous solutions under visible-light irradiation, the photocatalytic activities of four Bi2SiO5/g-C3N4 composites are compared and discussed.
2. Experimental
2.1. Materials
Na2SiO3 and ammonium oxalate (Osaka), Bi(NO3)3·5H2O, KI and urea (Katayama), CV dye (TCI), p-benzoquinone (Alfa Aesar), sodium azide and cetyltrimethylammonium bromide (CTAB) (Sigma-Aldrich), and isopropanol (Merck) were purchased and used without further purification. Reagent-grade sodium hydroxide, nitric acid, ammonium acetate, and HPLC-grade methanol were obtained from Merck.
2.2. Instrumentation and analytical methods
Field emission scanning electron microscopy-electron dispersive X-ray spectroscopy (FE-SEM-EDS) measurements were carried out using JEOL JSM-7401F machine at an acceleration voltage 15 kV. The Al-Kα radiation was generated at 15 kV. The X-ray diffraction (XRD) patterns were recorded on a MAC Science MXP18 equipped with Cu-Kα radiation, operating at 40 kV and 80 mA. The field-emission transmission electron microscopy (FE-TEM) images, selected area electron diffraction (SAED) patterns, high resolution transmission electron microscopy (HRTEM) images, and energy-dispersive X-ray spectra (EDS) were obtained using a JEOL-2010 machine with an accelerating voltage of 200 kV. Photoluminescence (PL) measurements were carried out on Hitachi F-7000. High resolution X-ray photoelectron spectroscopy (HRXPS) measurements were carried out using a ULVAC-PHI. The ultra-violet photoelectron spectroscopy (UPS) measurements were performed using a ULVAC-PHI XPS, PHI Quantera SXM. The Brunauer–Emmett–Teller (BET) specific surface areas of the samples (SBET) were measured with an automated system (Micrometrics Gemini) using nitrogen gas as the adsorbate at liquid nitrogen temperature.
2.3. Synthesis of Bi2SiO5/g-C3N4
Under atmospheric conditions, the g-C3N4 powder was synthesized by directly calcining melamine in a muffle furnace. In a typical synthesis run, 5 g melamine was placed in a semi-closed alumina crucible with a cover. The crucible was heated to 520 °C for 4 h with a heating rate of 10 °C min−1. After cooling to room temperature, g-C3N4 was produced in powder form.62 2 mmol Bi (NO3)3·5H2O and 1 mmol Na2SiO3 were first mixed in a 50 mL flask, followed by the addition 30 mL 4 M HNO3. With continuous stirring, 2 M NaOH were added dropwise to adjust the pH to 13. The solution was then stirred vigorously for 30 min and transferred into a 30 mL Teflon-lined autoclave, which was heated up to 150 °C for 24 h and then naturally cooled down to room temperature. Solution A (0.5 g g-C3N4 and 0.05 g CTAB followed by adding 10 mL ethylene glycerol) and solution B (0.5 g Bi2SiO5 followed by adding 10 mL ethylene glycerol and 0.07 g urea) were first mixed in a 50 mL flask. The solution was then stirred vigorously for 20 min and transferred into a 30 mL Teflon-lined autoclave, which was heated up to 150 °C for 4 h and then naturally cooled down to room temperature. The resulting solid precipitate was collected by filtration, washed with deionized water and methanol to remove any possible ionic species in the solid precipitate, and then dried at 60 °C overnight.
2.4. Photocatalytic experiments
The CV irradiation experiments were carried out on a stirred aqueous solution contained in a 100 mL flask; the aqueous suspension of CV (100 mL, 10 ppm) and the catalyst powder was placed in a Pyrex flask. The pH of the suspension was adjusted by adding either NaOH or HNO3 solution. The experiments were performed in the dark in order to examine the adsorption/desorption equilibrium. 10 mg of the photocatalyst was mixed with 100 mL of CV aqueous solution with a known initial concentration, in a 100 mL flask, and the mixture was shaken in an orbital shaker (100 rpm) at a constant temperature. The mixture was centrifuged at 3000 rpm in a centrifugation machine after batch sorption experiments so that the absorbance of CV could be determined at 580 nm by means of HPLC-PDA-ESI-MS. The concentrations of the solutions were determined using the linear regression equation. Prior to the irradiation, the suspension was magnetically stirred in the dark for ca. 30 min to establish an adsorption/desorption equilibrium between the CV and the catalyst surface. Irradiation was carried out using 150 W Xe arc lamps; the light intensity was fixed at 31.2 W m−2, and the reaction vessel was placed at 30 cm from the light source. At given irradiation time intervals, a 5 mL aliquot was collected and centrifuged to remove the catalyst. The supernatant was measured by HPLC-PDA-ESI-MS.
A series of quenchers was introduced to scavenge the relevant active species, in order to evaluate the effect of the active species during the photocatalytic reaction. O2˙−, ˙OH, h+, and 1O2 were studied by adding 1.0 mM benzoquinone (BQ, a quencher of O2˙−),63 1.0 mM isopropanol (IPA, a quencher of ˙OH),64 1.0 mM ammonium oxalate (AO, a quencher of h+),65 and 1.0 mM sodium azide (SA, a quencher of 1O2),66 respectively. The method was similar to the previous photocatalytic activity test.
3. Results and discussion
3.1. Characterization of Bi2SiO5/g-C3N4 composites
3.1.1. Phase structure. Fig. 1 shows the XRD patterns of the as-prepared samples; the patterns clearly show the existence of the Bi2SiO5 phase composite with g-C3N4. All the samples as-prepared contain the Bi2SiO5 phase (JCPDS 01-075-1483) and the g-C3N4 (JCPDS 87-1526) phase. Fig. 2(a)–(d) display that the 10 wt% Bi2SiO5/g-C3N4 samples are composed of different-sized layers, consistent with the TEM observations. In addition, the EDS spectrum shows that the sample contains the elements of Bi, Si, O, C, and N. The HRTEM image shows that one set of different lattice images is found with a d-spacing of 0.372 nm, corresponding to the (310) plane of Bi2SiO5, which is in good agreement with the XRD results. The results suggest that the Bi2SiO5/g-C3N4 phases have been formed in the composites, which is favorable for the separation of photoinduced carriers, yielding high photocatalytic activities. Fig. 2(e)–(h) show the irregular edge morphology, which is consistent with the reference sample.67 The results suggest that the g-C3N4 phases have been formed.
 |
| | Fig. 1 XRD patterns of the as-prepared Bi2SiO5/g-C3N4 samples with different weight percentages (%), temp. = 150 °C, pH = 13, and time = 8 h. | |
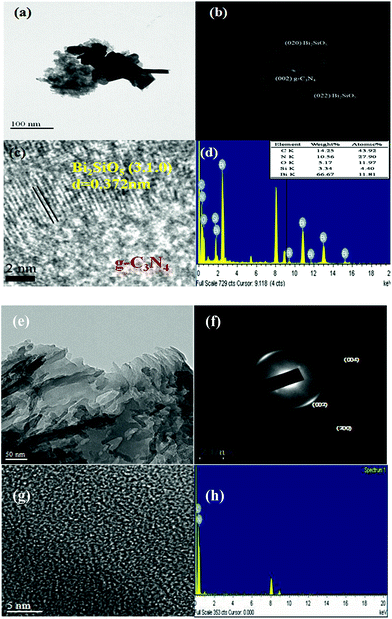 |
| | Fig. 2 FE-TEM and EDS of (a)–(d) as-prepared 10 wt% Bi2SiO5/g-C3N4 and (e)–(h) pure g-C3N4. | |
FT-IR spectra are shown in Fig. 3. Based on previously reports,68,69 the peaks located at around 430 cm−1, 570 cm−1, 857 cm−1, 946 cm−1 and 1030 cm−1 belong to the stretching vibration mode of Bi–O bonds, SiO44− groups, Bi–O–Si bonds, isolated SiO56− groups and Si–O bonds, respectively. Relative to pure g-C3N4, the peaks at 1252, 1326, 1420, 1572, and 1640 cm−1 correspond to the typical stretching modes of the CN heterocycles.70,71 In addition, the characteristic breathing mode of the triazine units at 811 cm−1 is observed.72 These results agree with those of the XRD and TEM experiments.
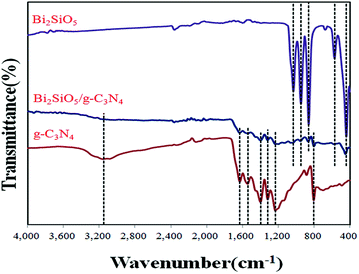 |
| | Fig. 3 FTIR spectra of as-prepared Bi2SiO5, g-C3N4, and 10 wt% Bi2SiO5/g-C3N4. | |
The proposed processes for the formation of Bi2SiO5/g-C3N4 composites are described in eqn (1)–(4). The results demonstrate a series of changes in the compounds prepared under different hydrothermal conditions, described as Bi2Si3O9 → Bi4Si3O12 → Bi2SiO5. By controlling the pH of the hydrothermal reaction, different compositions of bismuth silicates are obtained as follows:
| | |
2Bi3+ + 3SiO32− → Bi2Si3O9(s)
| (1) |
| | |
2Bi2Si3O9(s) + 6OH− → Bi4Si3O12(s) + 3SiO32− + 3H2O
| (2) |
| | |
Bi4Si3O12(s) + 2OH− → 2Bi2SiO5(s) + SiO32− + H2O
| (3) |
| | |
Bi2SiO5(s) + g-C3N4 → Bi2SiO5/g-C3N4(s)
| (4) |
3.1.2. X-ray photoelectron spectroscopy analysis. Fig. 4 presents the C 1s, N 1s, O 1s, Bi 4f, and Si 2p XPS spectra of the Bi2SiO5/g-C3N4 composites. Observation of the transition peaks involving the O 1s, C 1s, N 1s, Bi 4f, and Si 2p orbitals identifies that the catalysts are composed of Bi, Si, O, C, and N. Fig. 4(b) shows the high resolution C 1s spectrum of Bi2SiO5/g-C3N4 composites. There are mainly two carbon species displayed in the C 1s spectra. One peak (283.8 eV) is attributed to the sp2 C–C bond, and the other (287.25 eV), to the sp2-hybridized carbon in the N-containing aromatic ring (N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N). The latter is indicated as the major carbon species in polymeric g-C3N4.71 In Fig. 4(c), three peaks are deconvoluted for the N 1s spectra. The highest peak centering at 398.5 eV is assigned to the sp2-hybridized nitrogen involved in triazine rings (C–NC), whereas the peak at 401.0 eV corresponds to the tertiary nitrogen N–(C)3 groups. Both of them, together with sp2-hybridized carbon (N–CN, 287.9 eV), compose the heptazine heterocyclic ring units, constructing the basic substructure units of g-C3N4 polymers. The weak peak at 404.4 eV is attributed to charging effects or positive charge localization in the heterocycles.72 The asymmetric O 1s peak shown in Fig. 4(d) can be split by using the XPS peak-fitting program. The peak at 530.5 eV is assigned to the external –OH groups or the water molecules adsorbed on the surface, and the other O 1s peak appearing at 528.8 eV corresponds to lattice oxygen atoms in the Bi2SiO5.73 The characteristic binding energy value of 158.2 eV for Bi 4f7/2 (Fig. 4(e)) shows a trivalent oxidation state for bismuth. An additional spin–orbit doublet with the binding energy of 155.5 eV for Bi 4f7/2 is also revealed in all samples, suggesting that certain parts of bismuth exist in the (+3 − x) valence state. This shows that the trivalent bismuth is partially reduced to the lower valence state by the hydrothermal method. A similar chemical shift of approximately 2.7 eV for Bi 4f7/2 was also published by Liao et al.74 They summarized that the Bi(+3−x) formal oxidation state could most probably be ascribed to the sub-stoichiometric forms of Bi within the Bi2O2 layer, and the formation of the low oxidation state results in oxygen vacancies in the crystal lattice. However, it is supposed in this study that the Bi(+3−x) formal oxidation state could most likely be ascribed to the sub-stoichiometric forms of Bi at the outer sites of the particles, and the formation of the low oxidation state results in oxygen vacancies in the crystal surface, revealing that the main chemical states of the bismuth element in the samples are not trivalent. From Fig. 4(f), the binding energy of 101.0 eV is attributed to Si 2p3/2, which points to Si in the tetravalent oxidation state.
N). The latter is indicated as the major carbon species in polymeric g-C3N4.71 In Fig. 4(c), three peaks are deconvoluted for the N 1s spectra. The highest peak centering at 398.5 eV is assigned to the sp2-hybridized nitrogen involved in triazine rings (C–NC), whereas the peak at 401.0 eV corresponds to the tertiary nitrogen N–(C)3 groups. Both of them, together with sp2-hybridized carbon (N–CN, 287.9 eV), compose the heptazine heterocyclic ring units, constructing the basic substructure units of g-C3N4 polymers. The weak peak at 404.4 eV is attributed to charging effects or positive charge localization in the heterocycles.72 The asymmetric O 1s peak shown in Fig. 4(d) can be split by using the XPS peak-fitting program. The peak at 530.5 eV is assigned to the external –OH groups or the water molecules adsorbed on the surface, and the other O 1s peak appearing at 528.8 eV corresponds to lattice oxygen atoms in the Bi2SiO5.73 The characteristic binding energy value of 158.2 eV for Bi 4f7/2 (Fig. 4(e)) shows a trivalent oxidation state for bismuth. An additional spin–orbit doublet with the binding energy of 155.5 eV for Bi 4f7/2 is also revealed in all samples, suggesting that certain parts of bismuth exist in the (+3 − x) valence state. This shows that the trivalent bismuth is partially reduced to the lower valence state by the hydrothermal method. A similar chemical shift of approximately 2.7 eV for Bi 4f7/2 was also published by Liao et al.74 They summarized that the Bi(+3−x) formal oxidation state could most probably be ascribed to the sub-stoichiometric forms of Bi within the Bi2O2 layer, and the formation of the low oxidation state results in oxygen vacancies in the crystal lattice. However, it is supposed in this study that the Bi(+3−x) formal oxidation state could most likely be ascribed to the sub-stoichiometric forms of Bi at the outer sites of the particles, and the formation of the low oxidation state results in oxygen vacancies in the crystal surface, revealing that the main chemical states of the bismuth element in the samples are not trivalent. From Fig. 4(f), the binding energy of 101.0 eV is attributed to Si 2p3/2, which points to Si in the tetravalent oxidation state.
 |
| | Fig. 4 High resolution XPS spectra of the Bi2SiO5, g-C3N4 and 10 wt% Bi2SiO5/g-C3N4 composite: (a) total survey, (b) C 1s, (c) N 1s, (d) O 1s, (e) Bi 4f, and (f) Si 2p. | |
3.1.3. Morphological structure and composition. Fig. 5 shows the FESEM images of the Bi2SiO5, g-C3N4, Bi2SiO5/g-C3N4 composites at high magnification. The Bi2SiO5, g-C3N4, and Bi2SiO5/g-C3N4 samples show square-plates, nanosheets, and bulk morphology, respectively. The SEM-EDS and TEM-EDS results demonstrate that the main elements within these samples are carbon, nitrogen, oxygen, silicon, and bismuth, shown in Fig. 2 and 5. Based on the above results, Bi2SiO5/g-C3N4 composites could be selectively synthesized through a controlled hydrothermal method.
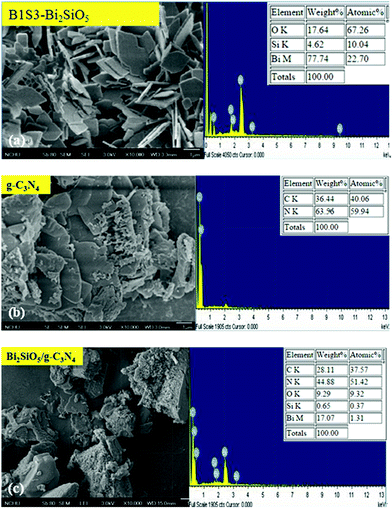 |
| | Fig. 5 FE-SEM and EDS of the as-prepared (a) Bi2SiO5, (b) g-C3N4, and (c) 10 wt% Bi2SiO5/g-C3N4. | |
3.1.4. Optical absorption properties. As shown in Fig. 6 for DR-UV of the Bi2SiO5/g-C3N4 composites, the absorption edge of the pure g-C3N4 is at about 482.5 nm, which originates from its band gap of 2.38 eV and is consistent with the reported results.74 Pure g-C3N4 and Bi2SiO5 absorb only a small amount of visible light. The Eg value of Bi2SiO5/g-C3N4 was determined from a plot of (αhν)1/2 vs. energy (hν), which was calculated as 2.46 eV.
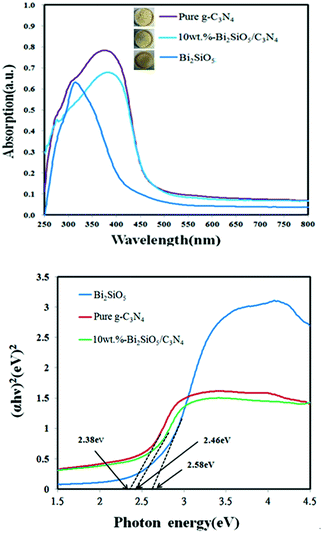 |
| | Fig. 6 DRS patterns of the as-prepared Bi2SiO5, g-C3N4, and 10 wt% Bi2SiO5/g-C3N4. | |
3.1.5. BET and adsorption–desorption isotherm. The 10 wt% Bi2SiO5/g-C3N4 composite has the middle SBET value and the highest pore volume (Table 1). However, the results from Table 2 show that the 10 wt% Bi2SiO5/g-C3N4 sample, which shows the middle SBET value, does represent the highest photocatalytic activity (k = 1.1257 × 10−1 h−1) among the samples, suggesting that the changes in the photocatalytic activity result from Bi2SiO5/g-C3N4 composites.
Table 1 Physical and chemical properties of the as-prepared samples
| Sample |
Specific surface area (m2 g−1) |
Pore volume (cm3 g−1) |
Pore diameter (nm) |
| Bi2SiO5 |
1.54 |
0.012 |
37.60 |
| g-C3N4 |
17.60 |
0.1975 |
28.54 |
| 10 wt% Bi2SiO5/g-C3N4 |
8.8108 |
0.112 |
37.85 |
Table 2 The pseudo-first-order rate constants for the degradation of CV with Bi2SiO5/g-C3N4 photocatalysts under visible light irradiation
| |
k (h−1) |
R2 |
| Pure-g-C3N4 |
0.0419 |
0.9892 |
| 1 wt% Bi2SiO5/g-C3N4 |
0.0814 |
0.9794 |
| 5 wt% Bi2SiO5/g-C3N4 |
0.0857 |
0.9580 |
| 7 wt% Bi2SiO5/g-C3N4 |
0.1104 |
0.9549 |
| 10 wt% Bi2SiO5/g-C3N4 |
0.1257 |
0.9745 |
| 20 wt% Bi2SiO5/g-C3N4 |
0.0360 |
0.9997 |
| 50 wt% Bi2SiO5/g-C3N4 |
0.0332 |
0.9783 |
| Bi2SiO5 |
0.0256 |
0.9701 |
Fig. 7 shows the nitrogen adsorption–desorption isotherm curves of Bi2SiO5, g-C3N4 and Bi2SiO5/g-C3N4. The isotherms of g-C3N4 are close to Type IV with a hysteresis loop at a high relative pressure between 0.6 and 1.0.75 The isotherms of Bi2SiO5 and Bi2SiO5/g-C3N4 are close to Type III, with hysteresis loops at a high relative pressure between 0.6 and 1.0. The shape of the hysteresis loop is close to Type H3, suggesting the existence of slit-like pores, generally formed by the aggregation of plate-like particles, which is consistent with the self-assembled nanoplate-like morphology of the samples. This result is consistent with the image results of FE-SEM and TEM, showing that self-assembled nanosheets or nanoplates result in the formation of hierarchical architectures.
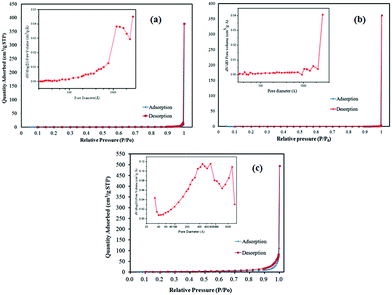 |
| | Fig. 7 N2 adsorption–desorption isotherms and pore size distribution of (a) Bi2SiO5, (b) g-C3N4, and (c) 10 wt% Bi2SiO5/g-C3N4. | |
3.2. Photocatalytic activity
The degradation efficiency as a function of reaction time is illustrated in Fig. 8(a). The removal efficiency is significantly enhanced in the presence of 10 wt% Bi2SiO5/g-C3N4. After irradiation for 48 h, the 10 wt% Bi2SiO5/g-C3N4 exhibits superior photocatalytic performance, with CV removal efficiency of up to 99%. To further understand the reaction kinetics of CV degradation, the apparent pseudo-first-order model76 expressed by ln(Co/C) = kt equation was applied in the experiments. Via the first-order linear fit of the data shown in Fig. 8(b) and Table 2, the k value of 10 wt% Bi2SiO5/g-C3N4 was obtained as the maximum degradation rate of 1.257 × 10−1 h−1 using the first-order linear fit of the data, which is much higher than that of the other composites; the 10 wt% Bi2SiO5/g-C3N4 composite is a much more effective photocatalyst than the others synthesized in this study. Comparison of rate constants of different photocatalysts is shown in Table 2. The order of the rate constants is 10 wt% Bi2SiO5/g-C3N4 > 7 wt% Bi2SiO5/g-C3N4 > 5 wt% Bi2SiO5/g-C3N4 > 1 wt% Bi2SiO5/g-C3N4 > 20 wt% Bi2SiO5/g-C3N4 > 50 wt% Bi2SiO5/g-C3N4. The photocatalytic activity of the 10 wt% Bi2SiO5/g-C3N4 heterojunctions reached the maximum rate constant of 1.257 × 10−1 h−1, which is 5 times higher than that of Bi2SiO5 and 3 times higher than that of g-C3N4. Thus, the Bi2SiO5/g-C3N4 composites may also play a role in enhancing the photocatalytic activity.
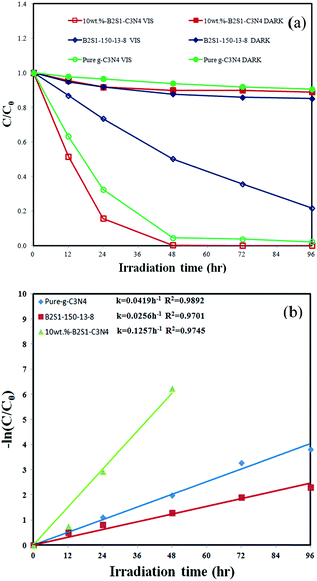 |
| | Fig. 8 Photodegradation of CV as a function of irradiation time, using the as-prepared Bi2SiO5, g-C3N4, and 10 wt% Bi2SiO5/g-C3N4. | |
The durability of the 10 wt% Bi2SiO5/g-C3N4 composite was evaluated by recycling the used catalyst. After each cycle, the catalyst was collected by centrifugation. No apparent loss was observed in the photocatalytic activity when CV was removed in the 3rd cycle; even during the fifth run, the decline in the photocatalytic activity was 3.5% (Fig. 9(a)). The used 10 wt% Bi2SiO5/g-C3N4 composite was also examined by XRD and no detectable difference was observed between the as-prepared and the used samples (Fig. 9(b)); hence, the 10 wt% Bi2SiO5/g-C3N4 composite has good photostability.
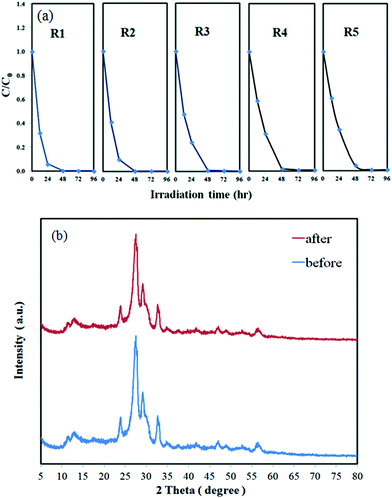 |
| | Fig. 9 (a) Cycling runs in the photocatalytic degradation of CV in the presence of 10 wt% Bi2SiO5/g-C3N4, (b) XRD of the sample powder before and after the degradation reaction. | |
As is known, the photocatalysts become excited and generate electron–hole pairs directly after illumination in the photocatalytic process. Moreover, the photocatalytic performance mainly depends on the recombination rate or the lifetime of the photo-generated electron–hole pairs. The faster recombination occurs, the less time is required for the chemical reactions. Therefore, PL spectra are utilized for investigating the recombination rates of the photogenerated electron–hole pairs.77 To investigate the separation capacity of the photogenerated carriers in the heterostructures, the PL spectra of g-C3N4, Bi2SiO5, and 10 wt% Bi2SiO5/g-C3N4 were obtained and the results are shown in Fig. 10. A strong emission peak around 458 nm appears for the as-prepared samples, which could have been derived from the direct electron–hole recombination of band transitions. However, the characteristic emission peak of lowest intensity, around 458 nm for the 10 wt% Bi2SiO5/g-C3N4, indicates that the recombination of photogenerated charge carriers is greatly inhibited. The efficient separation of charge could increase the lifetime of charge carriers and enhance the efficiency of interfacial charge transfer to the adsorbed substrates, thus improving the photocatalytic activity.78,79 The lowest relative PL intensities of 10 wt% Bi2SiO5/g-C3N4 composites, as shown in Fig. 10, suggest that they possess the lowest recombination rate of electron–hole pairs, resulting in their higher photocatalytic activity, as shown in Fig. 8(b). The PL results confirm the importance of the heterojunction in hindering the recombination of electrons and holes and explain the reason for the increasing photocatalytic performance of 10 wt% Bi2SiO5/g-C3N4 composites.
 |
| | Fig. 10 PL spectra of as-prepared Bi2SiO5, g-C3N4, and 10 wt% Bi2SiO5/g-C3N4. | |
It can be assumed that the enhanced photocatalytic activities of 10 wt% Bi2SiO5/g-C3N4 composites could be ascribed to the formation of the heterojunction. In the absence of photocatalysts, CV could not be degraded under visible-light irradiation. The superior photocatalytic ability of the 10 wt% Bi2SiO5/g-C3N4 may be ascribed to its efficient utilization of visible light and the high separation efficiency of the electron–hole pairs within its composites.
As shown in Table 3, graphitic carbon nitride composites have received remarkable interest in recent years because of their suitable band gaps, stability, and relatively superior photocatalytic activities. It is found that the graphitic carbon nitride composites show higher photocatalytic activities than g-C3N4 for the photocatalytic degradation of rhodamine B (or methyl blue, methyl orange, crystal violet).80–89
Table 3 Photocatalytic properties of composite photocatalysts under visible light irradiation
| Composite photocatalyst |
Mass fraction of g-C3N4 (wt%) |
Dye |
Photocatalytic activity |
Reference photocatalyst/photocatalytic activity |
Enhancement factor |
Reference |
| BiOI/g-C3N4 |
15 |
Rhodamine B |
90% decomposition in 30 min |
BiOI: 26.3% for rhodamine B |
3.8 |
80 |
| Bi5O7I/g-C3N4 |
30 |
Rhodamine B |
90% decomposition in 2 h |
g-C3N4: 6.5% |
15.3 |
81 |
| Bi5O7I: 34.5% |
2.9 |
| SrFeO3−x/g-C3N4 |
96 |
Crystal violet |
90% decomposition in 24 h |
g-C3N4: 20.8% |
4.8 |
82 |
| WO3/g-C3N4 |
90.3 |
Methylene blue |
97% decomposition in 2 h |
g-C3N4: 81%, 3 h |
1.2 |
83 |
| WO3: 73%, 3 h |
1.3 |
| CeO2/g-C3N4 |
87 |
Methylene blue |
95% decomposition in 2 h |
g-C3N4: 75%, 3 h |
1.3 |
84 |
| CeO2: 28%, 3 h |
3.4 |
| Bi2WO6/g-C3N4 |
50 |
Methyl orange |
93% decomposition in 2 h |
g-C3N4: 31% |
3 |
85 |
| Bi2WO6: 0.6% |
155 |
| Ag3VO4/g-C3N4 |
40 |
Basic fuchsin |
95% decomposition in 2.5 h |
g-C3N4: 15% |
6.3 |
86 |
| Ag3VO4: 30% |
3.2 |
| BiOBr/g-C3N4 |
50 |
Rhodamine B |
93% decomposition in 30 min |
g-C3N4: 15% |
6.3 |
87 |
| BiOBr: 35% |
2.7 |
| Ag/g-C3N4 |
Ag: 0.1 g |
Methyl orange |
74% decomposition |
g-C3N4: 70% |
1.3 |
88 |
| Ag/AgBr/g-C3N4 |
5 |
Methyl orange |
95.3% decomposition in 30 min |
g-C3N4: 2.5% |
38.1 |
89 |
| Ag/AgBr: 62.3% |
1.5 |
3.3. Photodegradation mechanisms of CV
In general, three possible reaction pathways are assumed to be involved in the photodegradation of organic compounds by a photocatalyst: (i) photocatalysis, (ii) photolysis, and (iii) dye photosensitization.90 In the photolysis process, a photoinduced electron on the induced compound directly reacts with O2 to produce 1O2, which acts as an oxidant for the photolysis of the compound.91 In the experiments, CV degradation induced by photolysis under visible light in a blank experiment was not observable; CV is a structure-stable dye and the decomposition by the photolysis mechanism is negligible.
As people may know, various primary active species, such as HO˙, h+, O2−˙, H˙ and 1O2, could be generated during photocatalytic degradation reactions in the UV-vis/semiconductor systems.91,92 Dimitrijevic et al.92 proposed that water, dissociated both on the surface of TiO2 and in subsequent molecular layers, has a three-fold role as follows: (i) the stabilization of charges, preventing electron–hole recombination, (ii) electron acceptor, involving the formation of H atoms in a reaction of photo-generated electrons with protons on the surface to give –OH2+, and (iii) electron donor, water reacts with photo-generated holes to give ˙OH radicals.
Ma et al. revealed that O2−˙ was the main active species for NO oxidation to NO3− with TO2/g-C3N4 under visible and UV light.93 Cheng's group illustrated a typical Z-scheme photocatalyst being favorable for the production of O2−˙ and ˙OH reactive species.94 Jiang et al. revealed that the photogenerated h+ and O2−˙ were the main oxidative species for the degradation of methyl orange.95 The generation of O2−˙ could not only inhibit the recombination of photoinduced charge carriers, but also benefit the de-chlorination of chlorinated phenol derivatives. The hydroxyl radical HO· might only be obtained via an e− → O2−˙ → H2O2 → ˙OH route. Moreover, ˙OH radicals were obtained by multistep reduction O2−˙ in the system.78 Hong et al. reported that O2−˙ played a major role in the V2O5/g-C3N4 heterojunctions photocatalytic reactions of rhodamine B and tetracycline, rather than the ˙OH radical.96 According to earlier studies,94 the photocatalytic process was mainly governed by O2−˙, rather than by ˙OH, e− or h+. The CV photodegradation by BiOmXn/BiOpXq (X, Y = Cl, Br, I) under visible light was dominated by O2−˙ oxidation, with O2−˙ being the main active species and ˙OH and h+ being the minor active species.78,97 On the basis of the references presented above, it is proposed that the probability of forming ˙OH should be much lower than that for O2−˙; however, ˙OH is an extremely strong and nonselective oxidant, which leads to the partial or complete mineralization of several organic chemicals.
In order to evaluate the effect of the active species during the photocatalytic processes, a series of quenchers was used to scavenge the relevant active species. As shown in Fig. 11, the photocatalytic degradation of CV was slightly affected by the addition of AO, IPA and SA, while the degradation efficiency of BQ quenching evidently decreases, compared with that of no-quenching, indicating that ˙OH, h+ and 1O2 are the minor active species, whereas O2˙− is the major active species in the process of the photocatalytic degradation of CV.
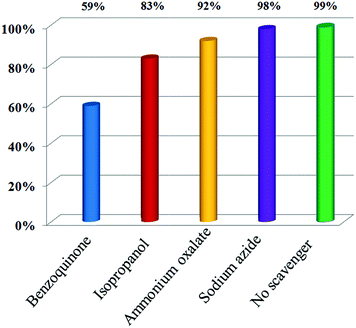 |
| | Fig. 11 Trapping of active species during the photocatalytic reaction using 10 wt% Bi2SiO5/g-C3N4. | |
In order to re-evaluate the effect of the active species during the photocatalytic reaction, EPR measurements were used to scavenge the relevant active species. From Fig. 12, not only were the six characteristic peaks of the DMPO–O2−˙ adducts observed, but the four characteristic peaks of DMPO–˙OH adducts (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1 quartet pattern) were also observed upon irradiation of the 10 wt% Bi2SiO5/g-C3N4 composite dispersion with visible light. Fig. 12(a) and (b) show that no EPR signal is observed when the reaction is performed in the dark, while the signals with intensities corresponding to the characteristic peaks of DMPO–˙OH and DMPO–O2−˙ adducts98 are observed during the reaction process under visible light irradiation, and the intensity gradually increases with prolonged reaction time, suggesting that O2˙− and ˙OH are formed as active species in the presence of 10 wt% Bi2SiO5/g-C3N4 composites and oxygen under visible light irradiation. Therefore, the quenching effects of different scavengers and EPR show that the reactive O2˙− plays the major role and ˙OH and h+ play minor roles in the photocatalytic degradation of CV.
:2:2:1 quartet pattern) were also observed upon irradiation of the 10 wt% Bi2SiO5/g-C3N4 composite dispersion with visible light. Fig. 12(a) and (b) show that no EPR signal is observed when the reaction is performed in the dark, while the signals with intensities corresponding to the characteristic peaks of DMPO–˙OH and DMPO–O2−˙ adducts98 are observed during the reaction process under visible light irradiation, and the intensity gradually increases with prolonged reaction time, suggesting that O2˙− and ˙OH are formed as active species in the presence of 10 wt% Bi2SiO5/g-C3N4 composites and oxygen under visible light irradiation. Therefore, the quenching effects of different scavengers and EPR show that the reactive O2˙− plays the major role and ˙OH and h+ play minor roles in the photocatalytic degradation of CV.
 |
| | Fig. 12 ESR spectra of (a) DMPO–˙OH and (b) DMPO–O2−˙ using 10 wt% Bi2SiO5/g-C3N4 dispersion in methanol solution under visible light irradiation. | |
Fan et al. revealed99 that Pt–TiO2 gathered less negative species on catalyst surfaces, which decreased reaction rates, than pure TiO2 did in an acidic environment. The ˙OH radical was produced subsequently, as also expressed in eqn (5)–(10).
| | |
O2−˙ + H+ + e− → HOO˙
| (5) |
| | |
HOO˙ + H2O → ˙OH + H2O2
| (6) |
| | |
H2O2 + e− → ˙OH + OH−
| (8) |
On the basis of the above experimental results, a proposed mechanism of degradation is illustrated in Fig. 13. Once the electron reaches the conduction band of Bi2SiO5, it induces the formation of active oxygen species, which cause the degradation of CV. It is clear that except for the photodegradation of CV by the route of Bi2SiO5/g-C3N4-mediated and photosensitized processes, another type of photocatalytic route accounts for the enhanced photocatalytic activity. In Fig. 13, both the photosensitized and photocatalytic processes proceed concurrently. However, in photosensitized and photocatalytic processes, O2˙− radicals are generated by the reaction of photogenerated and photosensitized electrons with oxygen gas on the photocatalyst surface, and ˙OH radicals are also generated by the reaction of O2˙− radicals with H+ ions, and holes h+ with OH− ions (or H2O). The ˙OH radical is produced subsequently, as expressed in eqn (5)–(10). These cycles continuously occur when the system is exposed to visible-light irradiation,78 and after several cycles of photo-oxidation, the degradation of CV by the produced oxidant species can be expressed by eqn (11) and (12):
| | |
CV + O2−˙/OH˙ → degraded compounds
| (11) |
| | |
CV+˙ + O2−˙/OH˙ → degraded compounds
| (12) |
 |
| | Fig. 13 The band structure diagram of Bi2SiO5/g-C3N4 and the possible charge separation processes. | |
In a visible-light-induced semiconductor system, hydroxylated compounds were also identified for the photocatalytic degradation of CV.78,97 In earlier reports,17,99–102 the N-dealkylation processes were preceded by the generation of a nitrogen-centered radical, and the destruction of the dye chromophore structure was preceded by the formation of a carbon-centered radical in the photocatalytic degradation of CV dye under UV or visible light irradiation. The reaction mechanisms for Bi2SiO5/g-C3N4-mediated photocatalytic processes proposed in this research should offer some insight for application to the decoloration of dyes.
The details of the separation and identification of intermediates have been provided in Fig. S1–S4 and Table S1 of ESI.† The details of the mechanisms of photocatalytic degradation have been shown in Fig. S5–S7 of the ESI.†
4. Conclusions
This is the first report on the synthesis of Bi2SiO5/g-C3N4 composites by a template-free hydrothermal method. The removal efficiency is significantly enhanced in the presence of 10 wt% Bi2SiO5/g-C3N4. The increased photocatalytic activities of Bi2SiO5/g-C3N4 could be attributed to the formation of the heterojunction between Bi2SiO5 and g-C3N4, which effectively suppresses the recombination of photo-generated electron–hole pairs. It can be concluded that the enhanced photocatalytic activities of Bi2SiO5/g-C3N4 materials could be due to the formation of the heterojunction. The quenching effects and EPR results illustrate that the reactive O2˙− plays the major role and ˙OH, h+ and 1O2 play minor roles in the CV degradation. This work is useful for the synthesis of Bi2SiO5/g-C3N4 and the photocatalytic degradation of the CV for future applications in environmental pollution and control.
Acknowledgements
This study was supported by the Ministry of Science and Technology of the Republic of China (MOST-104-2113-M-142-001).
Notes and references
- A. Kubacka, M. Fernández-García and G. Colón, Chem. Rev., 2012, 112, 1555–1614 CrossRef CAS PubMed.
- D. F. Duxbury, Chem. Rev., 1993, 93, 381–433 CrossRef CAS.
- B. P. Cho, T. Yang, L. R. Blankenship, J. D. Moody, M. Churchwell, F. A. Bebland and S. J. Culp, Chem. Res. Toxicol., 2003, 16, 285–294 CrossRef CAS PubMed.
- W. L. W. Lee, J. S. Lin, J. L. Chang, J. Y. Chen, M. C. Cheng and C. C. Chen, J. Mol. Catal. A: Chem., 2012, 361–362, 80–90 CrossRef CAS.
- F. Chen, P. Fang, Y. Gao, Z. Liu, Y. Liu and Y. Dai, Chem. Eng. J., 2012, 204–206, 107–113 CrossRef CAS.
- S. Nie, X. Zhao and B. Liu, RSC Adv., 2015, 5, 103386–103393 RSC.
- C. Y. Chen, J. T. Kuo, H. A. Yang and Y. C. Chung, Chemosphere, 2013, 92, 695–701 CrossRef CAS PubMed.
- F. Chen, P. Fang, Y. Gao, Z. Liu, Y. Liu and Y. Dai, Chem. Eng. J., 2012, 204–206, 107–113 CrossRef CAS.
- L. Ren, Y. Li, J. Hou, X. Zhao and C. Pan, ACS Appl. Mater. Interfaces, 2014, 6, 1608–1615 CAS.
- L. Q. Ye, J. Y. Liu, Z. Jiang, T. Y. Peng and L. Zan, Appl. Catal., B, 2013, 142–143, 1–7 CAS.
- S. Ameen, M. S. Akhtar, M. Nazim and H. S. Shin, Mater. Lett., 2013, 96, 228–232 CrossRef CAS.
- E. Ozkan, F. T. Ozkan, E. Allan and I. P. Parkin, RSC Adv., 2015, 5, 8806–8813 RSC.
- S. Girish Kumar and K. S. R. Koteswara Rao, RSC Adv., 2015, 5, 3306–3351 RSC.
- S. T. Huang, C. S. Lu, J. L. Chang, W. S. Huang and C. C. Chen, J. Taiwan Inst. Chem. Eng., 2014, 45, 1927–1936 CrossRef CAS.
- S. T. Huang, Y. R. Jiang, S. Y. Chou, Y. M. Dai and C. C. Chen, J. Mol. Catal. A: Chem., 2014, 391, 105–120 CrossRef CAS.
- K. Yu, S. Yang, C. Liu, H. Chen, H. Li, C. Sun and S. A. Boyd, Environ. Sci. Technol., 2012, 46, 7318–7326 CrossRef CAS PubMed.
- W. L. W. Lee, W. H. Chung, W. S. Huang, W. C. Lin, W. Y. Lin, Y. R. Jiang and C. C. Chen, J. Taiwan Inst. Chem. Eng., 2013, 44, 660–669 CrossRef CAS.
- V. L. Chandraboss, J. Kamalakkannan, S. Prabha and S. Senthilvelan, RSC Adv., 2015, 5, 25857–25869 RSC.
- Y. Wang, J. Duan, W. Li, S. Beecham and D. Mulcahy, J. Hazard. Mater., 2016, 303, 162–170 CrossRef CAS PubMed.
- S. Dutta, R. Sahoo, C. Ray, S. Sarkar, J. Jana, Y. Negishib and T. Pal, Dalton Trans., 2015, 44, 193–201 RSC.
- P. S. Kumar, M. Selvakumar, P. Bhagabati, B. Bharathi, S. Karuthapandian and S. Balakumar, RSC Adv., 2014, 4, 32977–32986 RSC.
- L. Chen, J. He, Q. Yuan, Y. W. Zhang, F. Wang, C. T. Au and S. F. Yin, RSC Adv., 2015, 5, 33747–33754 RSC.
- K. Yu, S. Yang, C. Liu, H. Chen, H. Li, C. Sun and S. A. Boyd, Environ. Sci. Technol., 2012, 46, 7318–7326 CrossRef CAS PubMed.
- W. Zhao, F. Liang, Z. M. Jin, X. B. Shi, P. H. Yin, X. R. Wang, C. Sun, Z. Q. Gao and L. S. Liao, J. Mater. Chem. A, 2014, 2, 13226–13231 CAS.
- W. Zhao, Y. Liu, Z. Wei, S. Yang, H. He and C. Sun, Appl. Catal., B, 2016, 185, 242–252 CrossRef CAS.
- S. Jin, Y. Li, H. Xie, X. Chen, T. Tian and X. Zhao, J. Mater. Chem., 2012, 22, 1469–1476 RSC.
- Y. Ide, F. Liu, J. Zhang, N. Kawamoto, K. Komaguchi, Y. Bandoa and D. Golberg, J. Mater. Chem. A, 2014, 2, 4150–4156 CAS.
- Y. Ma, H. Liu, Z. Han, L. Yang and J. Liu, J. Mater. Chem. A, 2015, 3, 14642–14650 CAS.
- F. Chen, Q. Yang, C. Niu, X. Li, C. Zhang and G. Zeng, RSC Adv., 2015, 5, 63152–63164 RSC.
- K. Laohhasurayotin and D. Viboonratanasri, Phys. Chem. Chem. Phys., 2013, 15, 9626–9635 RSC.
- C. Mondal, M. Ganguly, A. K. Sinha, J. Pal, R. Sahoo and T. Pal, CrystEngComm, 2013, 15, 6745–6751 RSC.
- S. Qin, W. Cai, X. Tang and L. Yang, Analyst, 2014, 139, 5509–5515 RSC.
- X. W. Lei, C. Y. Yue, L. J. Feng, Y. F. Han, R. R. Meng, J. T. Yang, H. Ding, C. S. Gao and C. Y. Wang, CrystEngComm, 2016, 18, 427–436 RSC.
- M. Shahid, I. E. Saliby, A. McDonagh, L. D. Tijing, J. H. Kim and H. K. Shon, J. Environ. Sci., 2014, 26, 2348–2354 CrossRef PubMed.
- A. K. Nayak, S. Lee, Y. Sohn and D. Pradhan, CrystEngComm, 2014, 16, 8064–8072 RSC.
- L. Li, X. Huang, T. Hu, J. Wang, W. Zhang and J. Zhang, New J. Chem., 2014, 38, 5293–5302 RSC.
- H. Cheng, B. Huang and Y. Dai, Nanoscale, 2014, 6, 2009–2026 RSC.
- L. Ye, J. Chen, L. Tian, J. Liu, T. Penga, K. Deng and L. Zan, Appl. Catal., B, 2013, 130–131, 1–7 CAS.
- Y. H. Liao, J. X. Wang, J. S. Lin, W. H. Chung, W. Y. Lin and C. C. Chen, Catal. Today, 2011, 174, 148–159 CrossRef CAS.
- J. A. Seabold and K. S. Choi, J. Am. Chem. Soc., 2012, 134, 2186–2192 CrossRef CAS PubMed.
- Z. Chen, H. Jiang, W. Jin and C. Shi, Appl. Catal., B, 2016, 180, 698–706 CrossRef CAS.
- W. Wei, Y. Dai and B. B. Huang, J. Phys. Chem. C, 2009, 113, 5658–5663 CAS.
- Y. Fei, S. Fan, R. Sun and M. Ishii, Prog. Cryst. Growth Charact., 2000, 40, 183–188 CrossRef.
- G. Corsmit, M. A. Van Driel and R. J. Elsenaar, J. Cryst. Growth, 1986, 75, 551–555 CrossRef CAS.
- R. Chen, J. Bi, L. Wu, W. Wang, Z. Li and X. Fu, Inorg. Chem., 2009, 48, 9072–9076 CrossRef CAS PubMed.
- H. W. Guo, X. F. Wang and D. N. Gao, Mater. Lett., 2012, 67, 280–282 CrossRef CAS.
- J. Lu, X. Wang, Y. Wu and Y. Xu, Mater. Lett., 2012, 74, 200–202 CrossRef CAS.
- H. Xie, C. Jia, Y. Jiang and X. Wang, Mater. Chem. Phys., 2012, 133, 1003–1005 CrossRef CAS.
- C. He and M. Gu, Scr. Mater., 2006, 55, 481–484 CrossRef CAS.
- J. Duan, Y. Liu, X. Pan, Y. Zhang, J. Yu, K. Nakajim and H. Taniguchi, Catal. Commun., 2013, 39, 65–69 CrossRef CAS.
- A. K. R. Police, S. Basavaraju, D. K. Valluri and S. Machiraju, J. Mater. Sci. Technol., 2013, 29, 639–646 CAS.
- S. S. Batool, S. Hassan, Z. Imran, M. A. Rafiq, M. Ahmad, K. Rasool, M. M. Chaudhry and M. M. Hasan, Catal. Commun., 2014, 49, 39–42 CrossRef CAS.
- D. Liu, W. Yao, J. Wang, Y. Liu, M. Zhang and Y. Zhu, Appl. Catal., B, 2015, 172–173, 100–107 CrossRef CAS.
- P. Wang, B. Huang, X. Qin, X. Zhang, Y. Dai, J. Wei and M. H. Whangbo, Angew. Chem., Int. Ed., 2008, 47, 7931–7933 CrossRef CAS PubMed.
- Z. Wan and G. Zhang, J. Mater. Chem. A, 2015, 3, 16737–16745 CAS.
- X. Liu, W. Wang, Y. Liu, B. Huang, Y. Dai, X. Qin and X. Zhang, RSC Adv., 2015, 5, 55957–55963 RSC.
- Y. Lei, X. Chen, C. Xu, Z. Dai and K. Wei, J. Catal., 2015, 321, 100–112 CrossRef CAS.
- Z. Zhao, Y. Sun and F. Dong, Nanoscale, 2015, 7, 15–37 RSC.
- C. Chang, L. Zhu, S. Wang, X. Chu and L. Yue, ACS Appl. Mater. Interfaces, 2014, 6, 5083–5093 CAS.
- H. Liu, Y. Su, Z. Chen, Z. Jin and Y. Wang, J. Hazard. Mater., 2014, 266, 75–83 CrossRef CAS PubMed.
- L. Sun, Y. Qi, C. J. Jia, Z. Jin and W. Fan, Nanoscale, 2014, 6, 2649–2659 RSC.
- B. P. Barbero and L. E. Cadus, Appl. Catal., A, 2002, 237, 263–273 CrossRef CAS.
- M. C. Yin, Z. S. Li, J. H. Kou and Z. G. Zou, Environ. Sci. Technol., 2009, 43, 8361–8366 CrossRef CAS PubMed.
- L. S. Zhang, K. H. Wong, H. Y. Yip, C. Hu, J. C. Yu, C. Y. Chan and P. K. Wong, Environ. Sci. Technol., 2010, 44, 1392–1398 CrossRef CAS PubMed.
- S. G. Meng, D. Z. Li, M. Sun, W. J. Li, J. X. Wang, J. Chen, X. Z. Fu and G. C. Xiao, Catal. Commun., 2011, 12, 972–975 CrossRef CAS.
- G. Li, K. H. Wong, X. Zhang, C. Hu, J. C. Yu, R. C. Y. Chan and P. K. Wong, Chemosphere, 2009, 76, 1185–1191 CrossRef CAS PubMed.
- G. Liao, S. Chen, X. Quan, H. Yu and H. Zhao, J. Mater. Chem., 2012, 22, 2721–2726 RSC.
- X. Feng, X. Qi, J. Li, L. Yang, M. Qiu, J. Yin, F. Lu and J. Zhong, Appl. Surf. Sci., 2011, 257, 5571–5575 CrossRef CAS.
- O. M. Bordun, J. Appl. Spectrosc., 1997, 64, 476–479 CrossRef CAS.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed.
- Y. Li, H. Zhang, P. Liu, D. Wang, Y. Li and H. Zhao, Small, 2013, 9, 3336–3344 CAS.
- J. Zhang, M. Zhang, G. Zhang and X. Wang, ACS Catal., 2012, 2, 2940–2948 Search PubMed.
- Y. R. Jiang, H. P. Lin, W. H. Chung, Y. M. Dai, W. Y. Lin and C. C. Chen, J. Hazard. Mater., 2015, 283, 787–805 CrossRef CAS PubMed.
- M. Xu, L. Han and S. Dong, ACS Appl. Mater. Interfaces, 2013, 5, 12533–12540 CAS.
- L. Lin, S. Yuan, J. Chen, L. Wang, J. Wan and X. Lu, Chemosphere, 2010, 78, 66–71 CrossRef CAS PubMed.
- A. Chatzitakis, C. Berberidou, I. Paspaltsis, G. Kyriakou, T. Sklaviadis and I. Poulios, Water Res., 2008, 42, 386–394 CrossRef CAS PubMed.
- K. Ishibashi, A. Fujishima, T. Watanabe and K. Hashimoto, Electrochem. Commun., 2000, 2, 207–210 CrossRef CAS.
- W. W. Dunn, Y. Aikawa and A. J. Bard, J. Am. Chem. Soc., 1981, 100, 3456–3459 CrossRef.
- S. T. Huang, Y. R. Jiang, S. Y. Chou, Y. M. Dai and C. C. Chen, J. Mol. Catal. A: Chem., 2014, 391, 105–120 CrossRef CAS.
- C. Chang, L. Zhu, S. Wang, X. Chu and L. Yue, ACS Appl. Mater. Interfaces, 2014, 6, 5083–5093 CAS.
- J. Di, J. Xia, S. Yin, H. Xu, L. Xu, Y. Xu, M. Hea and H. Li, J. Mater. Chem. A, 2014, 2, 5340–5351 CAS.
- H. P. Lin, C. C. Chen, W. W. Lee, Y. Y. Lai, J. Y. Chen, Y. Q. Chen and J. Y. Fu, RSC Adv., 2016, 6, 2323–2336 RSC.
- L. Y. Huang, H. Xu, Y. P. Li, H. M. Li, X. N. Cheng, J. X. Xia, Y. G. Xu and G. B. Cai, Dalton Trans., 2013, 42, 8606–8616 RSC.
- L. Y. Huang, Y. P. Li, H. Xu, Y. G. Xu, J. X. Xia, K. Wang, H. M. Li and X. N. Cheng, RSC Adv., 2013, 3, 22269–22279 RSC.
- Y. L. Tian, B. B. Chang, J. L. Lu, J. Fu, F. N. Xi and X. P. Dong, ACS Appl. Mater. Interfaces, 2013, 5, 7079–7085 CAS.
- S. M. Wang, D. L. Li, C. Sun, S. G. Yang, Y. Guan and H. He, Appl. Catal., B, 2014, 144, 885–892 CrossRef CAS.
- L. Q. Ye, J. Y. Liu, Z. Jiang, T. Y. Peng and L. Zan, Appl. Catal., B, 2013, 142–143, 1–7 CAS.
- Y. X. Yang, Y. N. Guo, F. Y. Liu, X. Yuan, Y. H. Guo, S. Q. Zhang, W. Guo and M. X. Huo, Appl. Catal., B, 2013, 142–143, 828–837 CrossRef CAS.
- J. Cao, Y. J. Zhao, H. L. Lin, B. Y. Xu and S. F. Chen, Mater. Res. Bull., 2013, 48, 3873–3880 CrossRef CAS.
- C. Nasr, K. Vinodgopal, L. Fisher, S. Hotchandani, A. K. Chattopadhyay and P. V. Kamat, J. Phys. Chem., 1996, 100, 8436–8442 CrossRef CAS.
- X. Xiao, R. Hao, M. Liang, X. Zuo, J. Nan, L. Li and W. Zhang, J. Hazard. Mater., 2012, 233–234, 122–130 CrossRef CAS PubMed.
- N. M. Dimitrijevic, B. K. Vijayan, O. G. Poluektov, T. Rajh, K. A. Gray, H. He and P. Zapol, J. Am. Chem. Soc., 2011, 133, 3964–3971 CrossRef CAS PubMed.
- J. Ma, C. Wang and H. He, Appl. Catal., B, 2016, 184, 28–34 CrossRef CAS.
- H. Cheng, J. Hou, O. Takeda, X. M. Guo and H. Zhu, J. Mater. Chem. A, 2015, 3, 11006–11013 CAS.
- D. Jiang, J. Li, C. Xing, Z. Zhang, S. Meng and M. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 19234–19242 CAS.
- Y. Hong, Y. Jiang, C. Li, W. Fan, X. Yan, M. Yan and W. Shi, Appl. Catal., B, 2016, 180, 663–673 CrossRef CAS.
- W. W. Lee, C. S. Lu, C. W. Chuang, Y. J. Chen, J. Y. Fu, C. W. Siao and C. C. Chen, RSC Adv., 2015, 5, 23450–23463 RSC.
- H. P. Lin, W. W. Lee, S. T. Huang, L. W. Chen, T. W. Yeh, J. Y. Fu and C. C. Chen, J. Mol. Catal. A: Chem., 2016, 417, 168–183 CrossRef CAS.
- H. J. Fan, C. S. Lu, W. L. W. Lee, M. R. Chiou and C. C. Chen, J. Hazard. Mater., 2011, 185, 227–235 CrossRef CAS PubMed.
- S. Ameen, M. S. Akhtar, M. Nazim and H. S. Shin, Mater. Lett., 2013, 96, 228–232 CrossRef CAS.
- Y. Li, S. Yang, C. Sun, L. Wang and Q. Wang, Water Res., 2016, 88, 173–183 CrossRef CAS PubMed.
- X. Li, G. Liu and J. Zhao, New J. Chem., 1999, 23, 1193–1196 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02299e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 












