
Insights into the macroporosity of freeze-cast hierarchical geopolymers
Abstract
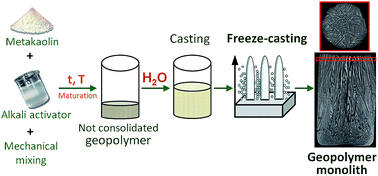
Geopolymer monoliths with controlled lamellar macroporosity and total porosity ranging from 60% to 70% were prepared by ice-templating a partially geopolymerized slurry. Both the maturation treatment of the starting mixture and the water specifically added for freeze-casting were tailored to modify both the geopolymerization and viscosity of the slurry, and, consequently, its freezing behavior, in order to optimize the final lamellar architecture. Following a room temperature maturation treatment, a 50% water content added for freezing developed thick lamellae and wide pores. A lower water content (30%) and curing at 80 °C after maturation at room temperature (for both 50% and 30% H2O) was conducive to a narrow lamellar pore width distribution in the 30–130 μm range. However, the consumption of water due to geopolymerization in samples cured at 80 °C led to a decreased length and thickness of the lamellae. Lastly, the interparticle meso- and macropores (0.003 to 1 μm) within the geopolymer lamellae were only slightly modified by the maturation treatment.
 Please wait while we load your content...
Please wait while we load your content...

