
DNA photo-cleaving agents in the far-red to near-infrared range – a review
Ziyi Li
and
Kathryn B. Grant
*
Department of Chemistry, Georgia State University, P.O. Box 3965, Atlanta, GA 30302-3965, USA. E-mail: kbgrant@gsu.edu; Fax: +1 404 413-5505; Tel: +1 404 413-5522
First published on 26th February 2016
Abstract
Photosensitizing agents that oxidatively cleave DNA are important to photodynamic therapy (PDT), an emerging treatment option for patients diagnosed with age-related macular degeneration, pre-cancerous conditions such as Barrett's esophagus, and localized cancers that include inoperable neoplasms. Excitation with low energy irradiation activates the PDT agents, causing spatially targeted, oxidative damage to DNA and other macromolecules in diseased cells. While most routinely used PDT photosensitizers rely on visible light sources that emit at wavelengths ≤689 nm, excitation that extends from the far-red into the near-infrared wavelength range is desirable. Due to low absorption by biogenic chromophores, light in this region has a greater penetration depth through tissue when compared to visible irradiation that is higher in energy. In the present review, we describe the development of new, long wavelength DNA photo-oxidizing agents. We have attempted to correlate the structural elements of the photosensitizers with their reported light-induced nuclease activities (λex ≥ 690 nm). Reaction pathways that lead to DNA cleavage are discussed, including 3O2 dependent Type I electron transfer and Type II energy transfer processes and anaerobic Type I hydrogen atom abstraction from deoxyribose. The summary discussion contained in this review is intended to contribute to the discovery of new phototherapeutic agents that are activated with far-red to near-infrared light.
 Ziyi Li | In 2014, Ziyi Li graduated with distinction from Georgia State University, receiving a B.S. degree in Chemistry with summa cum laude honors. She went on to obtain a chemistry M.S. degree after successfully defending her Masters thesis in the fall of 2015. Georgia State University presented Ziyi with awards for outstanding research at the undergraduate and Masters levels. Her current interests are focused on nucleic acids chemistry, cyanine dye chemistry, and nutrition. |
 Kathryn B. Grant | Kathryn B. Grant is a professor in the Department of Chemistry at Georgia State University. In 1980, she received a B.A. degree in Latin American Literature from New York University and in 1989, a B.S. degree in Chemistry from The State University of New York (College at Purchase). Her Ph.D. degree was obtained from Columbia University in 1997, studying the chemistry of blood and visual pigments under Professor Koji Nakanishi. She then carried out postdoctoral research in nucleic acids chemistry at CalTech. Kathy's research interests are currently focused on the design of metal complexes and chromophores that target disease-related macromolecules. |
1. Introduction
Many of the conventional anti-cancer drugs in clinical use exert their therapeutic effects by interacting with DNA (Scheme 1).1–9 Approximately 50% of cancer patients undergo treatment with a platinum-based therapeutic agent such as cisplatin (1), carboplatin (2), or oxaliplatin (3).4 The metal centers of these complexes coordinate to the nitrogen donor atoms of guanine bases, forming DNA mono-adducts as well as intra- and inter-strand cross-links.4,8 Additional examples of clinically approved anti-cancer agents that directly associate with nucleic acids include the DNA alkylating agents mitomycin C (4), mustine (5), cyclophosphamide (6), and dacarbazine (7).3,7 DNA intercalators such as actinomycin D (8), adriamycin (9), and mitoxantrone (10) act by either inhibiting transcription and/or DNA synthesis, or by inducing DNA strand breaks via topoisomerase inhibition.7 The therapeutic efficacy of the glycopeptide anti-tumor anti-biotic bleomycin A2 (11) is related to its ability to form DNA double-strand breaks in the presence of Fe(II) (or Cu(I)), O2, and a one-electron reducing agent.1,5,6,9 | ||
| Scheme 1 Conventional anti-cancer drugs that interact with DNA. | ||
Most conventional anti-cancer agents kill cells irrespective of whether they are normal or malignant. As a result, patients undergoing treatment can suffer from a range of adverse side effects including but not limited to bone marrow suppression, gastrointestinal distress, kidney, liver and lung damage, cardio toxicity, and permanent infertility.1,3,4,7,8,10 Still in clinical use, the nitrogen mustard mustine (5; HN2, bis(2-chloroethyl)methylamine, chloromethine, mechlorethamine, mustargen) was the first anti-cancer agent to have been approved by the United States Food and Drug Administration (FDA).10 Based on its potential to be used as a chemical weapon, mustine is currently classified as a Schedule 1 substance by the Chemical Weapons Convention.11 The frequency and severity of adverse side effects are major drawbacks associated with mustine and as well as many conventional anti-cancer agents.1,3,4,7,8,10
In order to reduce unfavorable drugs reactions and enhance positive treatment outcomes, researchers have long endeavored to design and implement new therapeutic strategies that selectively target diseased cells. Photodynamic therapy (PDT) is an emerging non-invasive treatment approach founded on the investigation of synthetic and naturally occurring photosensitizing agents.12–18 PDT is currently utilized in the treatment of age-related macular degeneration, pre-cancerous conditions such as actinic keratosis and Barrett's esophagus, and localized cancers including obstructing neoplasms for which surgery is not an option.12,14–17 In the PDT process, a chromophore called a photosensitizer (PS) is either introduced systemically or delivered directly to target tissue (Fig. 1). The photosensitizing agent is ideally non-toxic, but is activated to an excited singlet state (1PS*) upon exposure to light of a specific wavelength. The 1PS* excited state undergoes intersystem crossing to a long-lived triplet state (3PS*), which reacts with ground state triplet oxygen (3O2) to generate the short-lived reactive oxygen species (ROS) singlet oxygen (1O2) and hydroxyl radicals (˙OH) (Fig. 2).18 At the subcellular level, the photosensitizers can be taken up by different organelles including mitochondria, lysosomes, and nuclei.13–16 The 1O2 and ˙OH produced upon photo-activation have estimated, respective diffusion distances of 50–100 nm (ref. 19 and 20) and 0.8–6.0 nm,21 enabling them to inflict highly localized photo-oxidative damage to DNA, proteins, lipids, and other macromolecules that are encountered. Under ideal conditions, the spatially targeted cells undergo apoptosis or necrosis12–17,22 with minimal involvement of healthy surrounding tissues.12,15,23 This allows artificial control over the location and duration of anti-cancer treatment, contributing to high selectivity and promise of the PDT method to reduce the severe adverse side effects associated with conventional chemo-therapeutic agents.
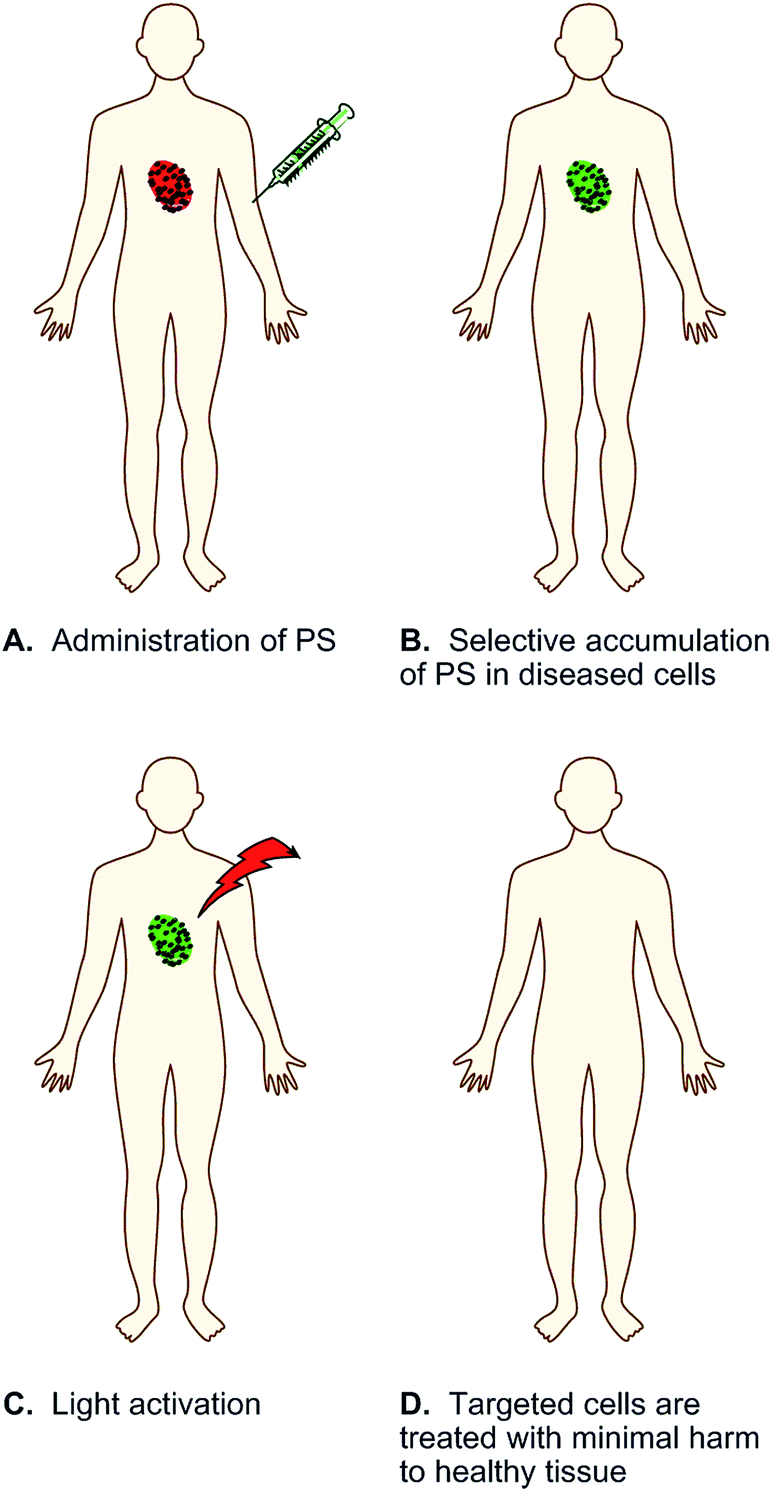 | ||
| Fig. 1 Schematic representation of photodynamic therapy (PDT), in which the therapeutic agent is a photosensitizer (PS) that is activated with low energy light. | ||
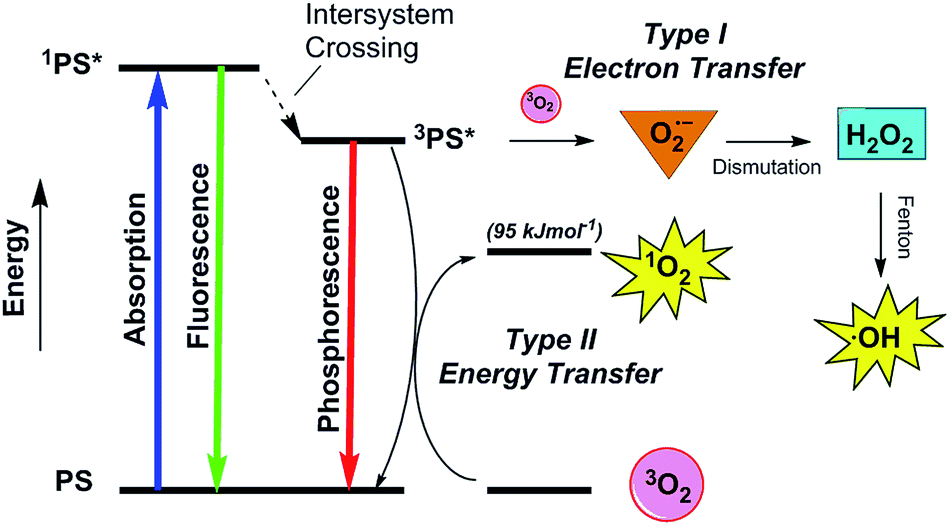 | ||
| Fig. 2 A simplified Jablonski diagram illustrating photosensitized production of Type I hydroxyl radicals (˙OH) and Type II singlet oxygen (1O2) from the excited triplet state (3PS*) of a photosensitizing agent (PS). | ||
In order to ensure efficient ROS production, a PDT photosensitizer should ideally possess high molar absorptivity and a first excited triplet state with a long lifetime (τT > 1 μs) and high quantum yield (ΦT > 0.4).18,24 Although photo-processes can take place between ground state triplet molecular oxygen (3O2) and a photosensitizer's first excited singlet state, reactions occurring from triplet states are generally more efficient.18,25,26 Oxidizing hydroxyl radicals (˙OH) are generated by a Type I photosensitization process in which the excited triplet state of the PS reacts with 3O2 by electron transfer to form superoxide anion radicals (O2˙−) (Fig. 2). The O2˙− radicals undergo spontaneous dismutation to generate hydrogen peroxide (H2O2), which then reacts with redox active metal ions to form hydroxyl radicals (˙OH) via Fenton-type chemistry.12,14,22,25 The ROS singlet oxygen (1O2) is produced in a Type II photosensitization reaction involving energy transfer from the sensitizer's triplet state to ground state triplet oxygen (3O2) (Fig. 2).12,14,18,22
An additional important factor that contributes to the level of ROS production in PDT is the energy of the photon used to activate a photosensitizing agent. Incident irradiation should ideally be capable of passing through spatially targeted areas far enough to cause significant oxidative damage to deeply embedded, diseased tissues. The absorption of ultra-violet and visible light by water, DNA, proteins, hemoglobin, melanin and other biogenic chromophores combined with increased water absorption and reduced light scattering at long wavelengths suggests that there is an optimal “optical window” of approximately 700 nm to 1200 nm for maximum transmittance of incident light.27 While the wavelength of the absorbed photon must be long enough to penetrate the living tissues deeply, Type II singlet oxygen can be efficiently generated only when the PDT photosensitizer has a triplet state energy that is equivalent to or higher than the excitation energy of 1O2 (95 kJ mol−1, ∼1270 nm; Fig. 2).18 When taking into consideration the standard energy gap of ∼63 kJ mol−1 between the first excited 1PS* and 3PS* states of many PDT systems, this corresponds to an absorption wavelength ≤750 nm.24,26,28 For photosensitizing agents with small singlet–triplet energy gaps (≤63 kJ mol−1), singlet oxygen can be produced at wavelengths up to ∼810 nm.28 In addition to energy transfer processes, the energy of an absorbed photon can activate a photosensitizing agent to undergo redox reactions. In order for light-sensitized superoxide anion radical production to occur, the reduction potential of the PS triplet state should be lower than the reduction potential of ground state triplet oxygen (E° (3O2/O2˙−) = −0.33 V at pH 7.0). When this criterion is met, ˙OH can be generated upon absorption of light in the ultra-violet, visible, and near-IR 700 nm to 900 nm wavelength regions.25,29
Interestingly, the majority of PDT photosensitizing agents in widespread clinical use are activated outside of the 700 nm to 1200 nm optical window for efficient light transmission through biological tissue. Porfimer sodium (12; Photofrin®) is currently the most commonly used FDA-approved drug mainly for Barrett's esophagus30 and for esophageal31 and endobronchial non-small cell lung cancers (Scheme 2).14,32,33 It consists of a mixture of porphyrin oligomers and is activated with a red light source emitting at 630 nm. Three additional photosensitizing agents have been approved by the FDA for routine clinical use.16 Activated at 689 nm, the monomeric porphyrin derivative verteporfin (13; Visudyne®) is an effective, palliative treatment for age-related macular degeneration, one of the leading causes of blindness in people over the age of 50.34 Used with light sources that emit at 417 nm and 635 nm respectively, the actinic keratosis medications δ-aminolevulinic acid (14; δ-ALA, Levulan®) and methyl aminolevulinate (15; MAL, Metvixia®) are metabolic precursors of photodynamically active protoporphyrin IX (16; PpIX).35 Additional porphyrinoid photosensitizing agents have been approved by international drug regulatory agencies.14,16 Photo-activated at 652 nm, the chlorin derivative temoporfin (17; Foscan®) is marketed in the European Union for treatment of squamous cell carcinoma of the head and neck.36 PDT with the chlorins talaporfin (18; Laserphyrin®) and Ce6-PVP (Photolon®), a complex of chlorin e6 (19) with low molecular polyvinylpyrrolidone, is carried out with a 660 nm light source.14 Talaporfin is utilized in Japan mainly for the treatment of lung cancer,33,37 while Ce6-PVP is employed in Russia primarily for cervical, lung, and skin tumors.38
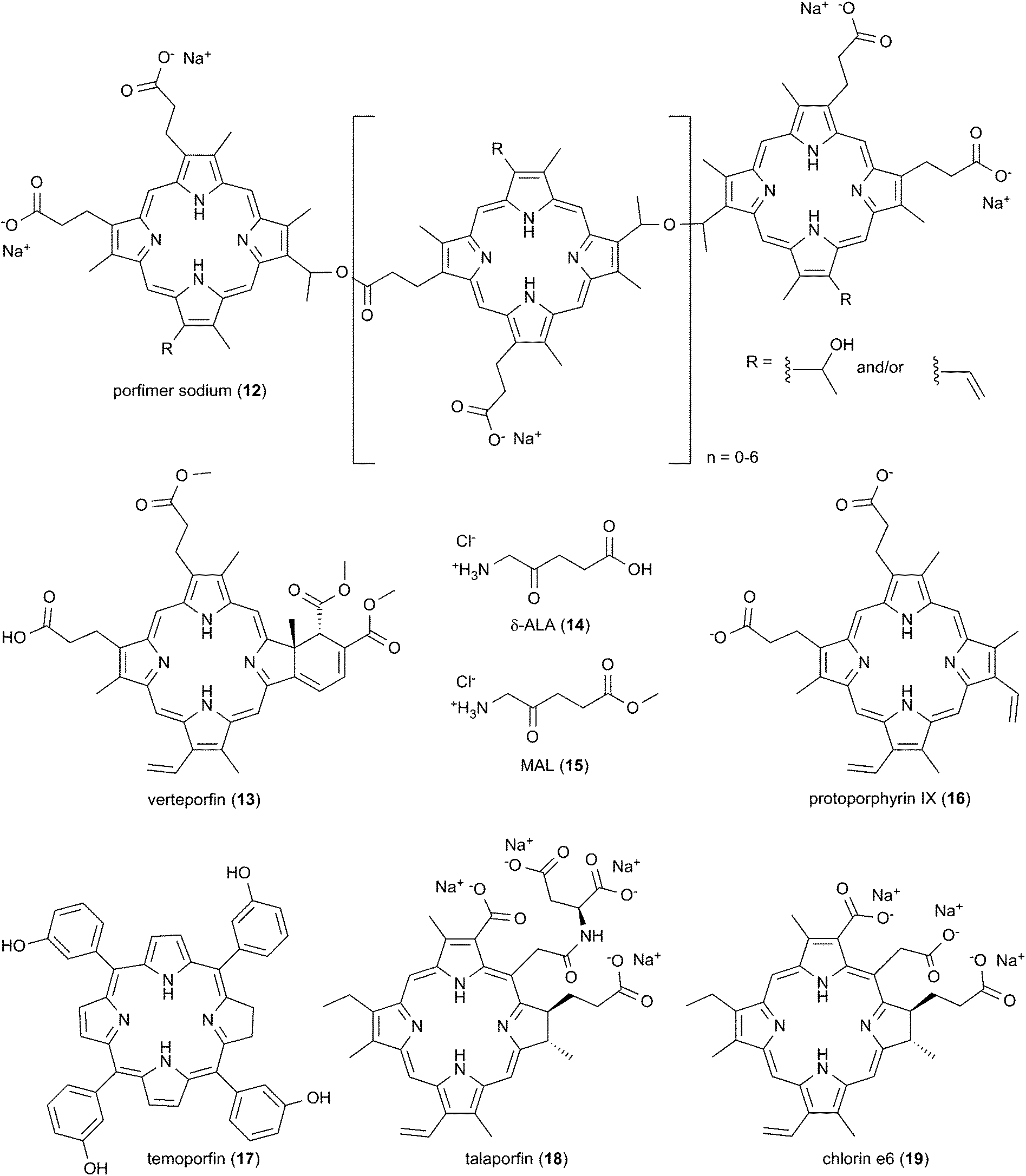 | ||
| Scheme 2 PDT photosensitizing agents in routine clinical use. | ||
Published meta analyses evaluating the effectiveness of PDT in clinical trials have found that systemic side effects are greatly reduced compared to therapeutic approaches involving conventional chemotherapeutic agents.14 The meta studies have reported high rates of complete response in the treatment of the early stage cancers: e.g., oral cavity squamous cell carcinoma (95%, porfimer sodium; 84–96%, temoporfin);39 superficial basal cell carcinoma (92%, porfimer sodium; 92%, δ-ALA; 97%, MAL);14 laryngeal carcinoma (92%, porfimer sodium);14 Tis/T1 primary head and neck tumors (85–95%, temoporfin);14,32,36 Tis/T1 lung cancer (72–85%, porfimer sodium; 85–94%, talaporfin);14,37 bladder cancer (74%, porfimer sodium);14 and esophageal cancer (44–87%, porfimer sodium).14,31 In the case of advanced disease, complete remission rates are generally lower: e.g., nodular basal cell carcinoma (71%, δ-ALA);14 recurrent head and neck tumors (66%, temoporfin);14,36 stage II/III head and neck tumors (13–38%, temoporfin);14 T2 esophageal cancer (28%, porfimer sodium); stage II/IV lung cancer (13%, porfimer sodium).14,33 As mentioned above, photosensitizing agents currently approved for routine clinical use maximally absorb at wavelengths under 690 nm. However, the effective penetration depth of light through tissue increases from ∼590 nm up to ∼840 nm.40 The average tissue penetration depth at 835 nm is approximately twice that at ∼630 nm, the wavelength used to activate porfimer sodium in photodynamic cancer therapy (Fig. 2).12,16,41 While it is clear that the efficacy of PDT is high in the case of early stage disease, it should be possible to improve the clinical outcomes obtained upon the treatment of larger, localized tumors by increasing the effective penetration depth of incident irradiation. This calls for the development of new photosensitizing agents that generate ROS in the far-red to near-infrared wavelength range.12,14,16,17
Many photosensitizers used in photodynamic therapy possess aromatic ring systems that have the potential to promote interactions with double-helical nucleic acids, either by intercalating in between base pairs and/or by binding within the DNA grooves. The DNA molecules in mitochondria and in the cell nucleus therefore constitute reasonable drug targets.42–47 While conventional anti-cancer agents such as cisplatin (1), mitomycin C (4), adriamycin (9), and bleomycin (11) modify DNA by alkylation and/or direct strand cleavage,1,3,4,7,8 porfimer sodium (12), verteporfin (13), talaporfin (18), and other PDT agents photocleave genomic DNA when irradiated in the presence of tissue culture and/or in circulating cells.48–53 The 1O2 and ˙OH produced upon photo-activation can cause direct strand breaks in nucleic acids.54,55 Several photosensitizing agents that cleave DNA in vitro localize in organelles where DNA is located, either in mitochondria44,56 and/or in the nuclei of tissue culture cells.42,43,45,46,57 Photo-activation of the sensitizers can cause photocytotoxicity in cancer cell lines with minimal dark toxicity and minimal toxicity to normal cells.45,46,57 For these reasons, the development of second-generation far-red to near-infrared PDT agents has involved the study of photosensitizing agents that cleave DNA.58–62
Over the past two decades, several excellent review papers on the topic of DNA photocleavage have been published.58,60–65 However, none of them has exclusively focused on photosensitizers that are activated with long wavelength light. Herein, we have attempted to organize, summarize, and analyze the key research studies recently published on this subject. Taking into consideration the excitation wavelengths of PDT agents in widespread clinical use (417 nm to 689 nm) and the deep penetration of light at long wavelengths, this review exclusively considers photosensitizers with DNA cleaving activity in far-red to near-IR light range (λex ≥ 690 nm). Among the selected, original papers, a majority of the reported compounds possess one or more paramagnetic metal ion centers and heterocyclic aromatic rings systems as ligands and/or DNA binding elements. Potential advantages of introducing metal ions into the chromophores are highlighted, including their ability to impart onto the photosensitizing agents flexible and varied geometries as well as d–d transition and/or charge transfer absorption bands that overlap with excitation wavelengths in the far-red to near-IR range. Several of the metal complexes described in this review are appended with chemical tags that facilitate selective photosensitizer uptake by target cancer cells46,57 and by organelles that induce an apoptotic response.44,56
This paper is basically divided into five sections according to photosensitizer type. Activation wavelengths appear in parenthesis. Photosensitizers incorporating metals include complexes containing the following metal ion centers: (i) Lu(III) (>700 nm);66 (ii) Cu(II) (694 nm to 755 nm);67–72 (iii) V(IV) (705 nm to 785 nm),42,44,46,56,57,73–81 and (iv) Fe(III) (785 nm).43 DNA photosensitization and cleavage by non-metalated organic dyes are discussed last.82–84 Included under this category are: (v) phenothiazine (710 nm) and cyanine (700 nm) photosensitizers,82,83 in addition to a carbazole chromophore (λmax = 419 nm) that is activated by 800 nm femtosecond two-photon irradiation.84 In the majority of studies covered in this review, photosensitizing agents generate high levels of DNA photocleavage by oxygen dependent pathways in which far-red to near-infrared irradiation sensitizes the production of DNA damaging Type I hydroxyl radicals (750 nm to 799 nm)42–44,46,56,57,72,74–78,80,81 and/or Type II singlet oxygen (705 nm to 753–799 nm).66,70,73,82 There are three published studies in which chromophores achieve quantitative DNA cleavage anaerobically through a Type I process that is proposed to involve hydrogen atom abstraction from deoxyribose (694 nm to 800 nm).69,71,84 In addition to absorbing long wavelength light that penetrates deeply through biological tissue, the latter compounds are of considerable interest due to their potential for extending the PDT approach to include the treatment of solid tumors that thrive under hypoxic conditions.20
2. Lutetium(III) texaphyrin
The first example of DNA photocleavage at an excitation wavelength ≥690 nm was reported by Sessler and co-workers in 1995.66 The photosensitizing agent used in these experiments consisted of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex of lutetium(III) and an “expanded porphyrin” ring system known as a texaphyrin (Scheme 3, Table 1). Compared to tetrapyrrolic porphyrins, texaphyrins have an additional coordinating nitrogen atom, affording a central core large enough to form stable, non-labile 1:1 complexes with large metal cations.85,86 In their experiments, Sessler and co-workers equilibrated lutetium(III) texaphyrin 20 (LuTx) with plasmid DNA and then irradiated the resulting solution with a xenon lamp fitted with a 700 nm cut off filter. Reaction products were easily visualized by conducting a plasmid DNA nicking assay similar to the schematic shown in Fig. 3. The experiment showed that the plasmid was photocleaved into nicked DNA in 93% yield when irradiated for 60 min in the presence of a 4 μM concentration of 20. Direct DNA strand breaks were produced, without requiring the addition of an external reducing agent or a base such as piperidine to induce cleavage at alkaline labile lesions. No cleavage was observed in parallel control reactions run in the dark. The activity of lutetium(III) texaphyrin 20 far surpassed the known porphyrin-based photonuclease meso-tetrakis(4-N-methylpyridyl)porphine (21, TMPyPH2), which produced little if any cleavage when reacted with DNA under identical conditions.66 While 20 has strong absorption in the near-infrared 700 nm to 800 nm region (εmax at 732 nm ∼42000 M−1 cm−1),16 TMPyPH2 exclusively absorbs light in the visible range (εmax at 586 nm ∼5900 M−1 cm−1).87 Lutetium(III) texaphyrin 20 has the added advantage possessing the “heavy atom” Lu(III) with the capacity to enhance intersystem crossing from the excited singlet state, thus facilitating triplet state sensitized ROS production. Whereas paramagnetic metals can shorten the lifetime of the excited triplet state of porphyrin-based photosensitizers, the metal ion center of 20 is diamagnetic (Lu(III) = [Xe] 4f14).25,85 In Sessler and co-workers' study, piperidine treatment of LuTx-sensitized photocleavage reactions revealed oxidative damage at guanine bases, a finding consistent with singlet oxygen production. The involvement of 1O2 was then confirmed by conducting nicking assays under a nitrogen atmosphere and in the presence of the singlet oxygen scavenger sodium azide.
:1 complex of lutetium(III) and an “expanded porphyrin” ring system known as a texaphyrin (Scheme 3, Table 1). Compared to tetrapyrrolic porphyrins, texaphyrins have an additional coordinating nitrogen atom, affording a central core large enough to form stable, non-labile 1:1 complexes with large metal cations.85,86 In their experiments, Sessler and co-workers equilibrated lutetium(III) texaphyrin 20 (LuTx) with plasmid DNA and then irradiated the resulting solution with a xenon lamp fitted with a 700 nm cut off filter. Reaction products were easily visualized by conducting a plasmid DNA nicking assay similar to the schematic shown in Fig. 3. The experiment showed that the plasmid was photocleaved into nicked DNA in 93% yield when irradiated for 60 min in the presence of a 4 μM concentration of 20. Direct DNA strand breaks were produced, without requiring the addition of an external reducing agent or a base such as piperidine to induce cleavage at alkaline labile lesions. No cleavage was observed in parallel control reactions run in the dark. The activity of lutetium(III) texaphyrin 20 far surpassed the known porphyrin-based photonuclease meso-tetrakis(4-N-methylpyridyl)porphine (21, TMPyPH2), which produced little if any cleavage when reacted with DNA under identical conditions.66 While 20 has strong absorption in the near-infrared 700 nm to 800 nm region (εmax at 732 nm ∼42000 M−1 cm−1),16 TMPyPH2 exclusively absorbs light in the visible range (εmax at 586 nm ∼5900 M−1 cm−1).87 Lutetium(III) texaphyrin 20 has the added advantage possessing the “heavy atom” Lu(III) with the capacity to enhance intersystem crossing from the excited singlet state, thus facilitating triplet state sensitized ROS production. Whereas paramagnetic metals can shorten the lifetime of the excited triplet state of porphyrin-based photosensitizers, the metal ion center of 20 is diamagnetic (Lu(III) = [Xe] 4f14).25,85 In Sessler and co-workers' study, piperidine treatment of LuTx-sensitized photocleavage reactions revealed oxidative damage at guanine bases, a finding consistent with singlet oxygen production. The involvement of 1O2 was then confirmed by conducting nicking assays under a nitrogen atmosphere and in the presence of the singlet oxygen scavenger sodium azide.
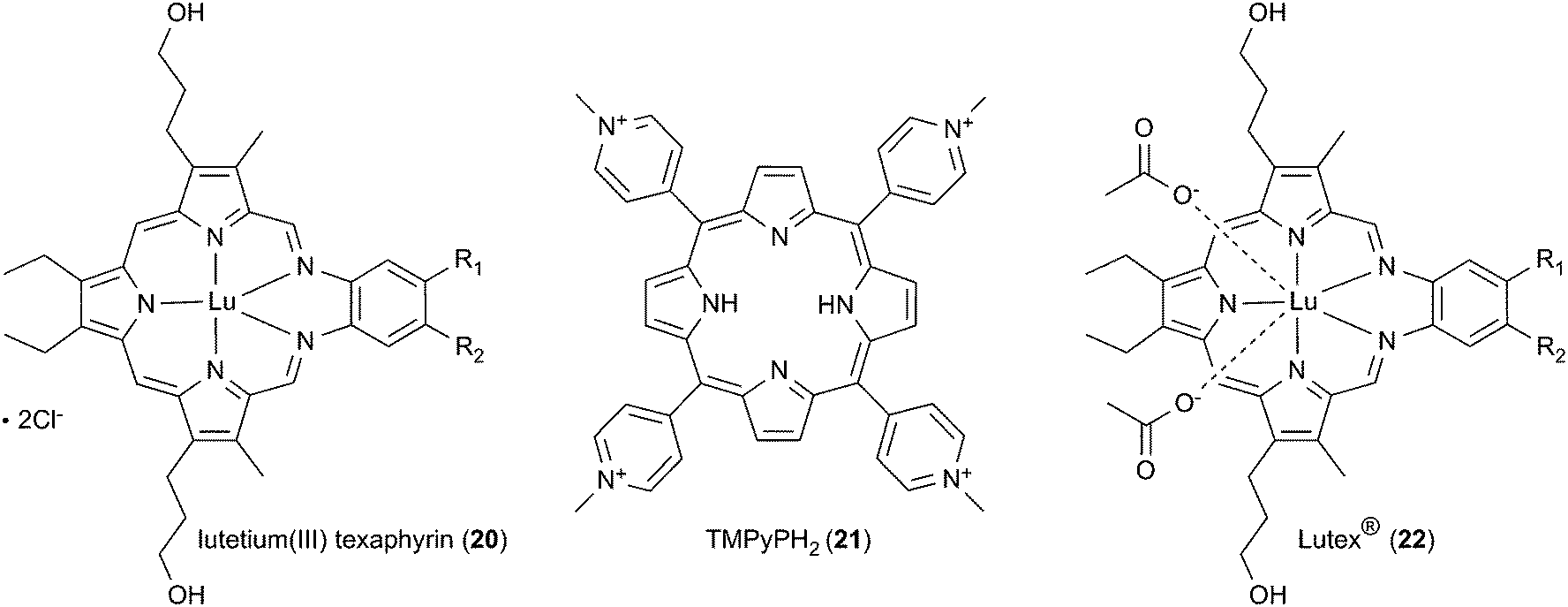 | ||
| Scheme 3 The structures of texaphyrins 20 and 22 compared to tetrapyrrolic porphyrin 21. R1 = R2 = OCH2CH2CH2OH for 20 and (OCH2CH2)3OCH3 for 22. | ||
| Photosensitizing agent | Plasmid DNA photocleavage reactions | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Description | λmaxb (nm) | εmaxc (M−1 cm−1) | Conc. (μM) | λexd (nm) | Time (min) | Yield (%) | Reactive speciese | Notes | |
| a Abbreviations: 2PA, two-photon absorption; dpq, dipyridoquinoxaline; dppz, dipyridophenazine; nap, naphthalimide; phen, 1,10-phenanthroline; n.r., not reported; TMBA, (3-bromopropyl)trimethylammonium bromide; TPP-ph, p-triphenylphosphonium methylphenyl bromide.b Local long wavelength λmax.c εmax at local long wavelength λmax.d DNA photocleavage excitation wavelength.e Reactive species were detected at DNA photocleavage excitation wavelength reported in preceding column, with exceptions noted in parenthesis. | |||||||||
| 20 (ref. 66) | Lu(III) texaphyrin | 732 | 42000 |
4 | ≥700 | 60 | 93 | 1O2 | High photocleavage; 22 tested in clinical trials |
| 23 (ref. 67) | Bis(dpq) Cu(II) complex | 673 | 70 | 50 | 694 | 120 | 98 | 1O2 (312 nm) | — |
| ˙OH (633 nm) | |||||||||
| 24 (ref. 68) | NSO donor Schiff base Cu(II) phen complex | 662 | 120 | 50 | 694 | 30 | 50 | 1O2 (603 nm) | Exhibits chemical nuclease activity |
| 25 (ref. 70) | L-Lysyl Cu(II) dppz complex | n.r. | n.r. | 80 | 735 | 40 | 96 | 1O2 | Possible chemical nuclease activity |
| 26 (ref. 69) | Binuclear NO donor Schiff base Cu(II) disulfide bridged complex | 640 | 300 | 400 | 694 | 240 | 98 | 1O2 (312 nm) | Anaerobic photocleavage at 633 nm suggested RS˙− involvement |
| ˙OH (633 nm) | |||||||||
| 27 (ref. 71) | Binuclear dpq Cu(II) disulfide bridged complex | 661 | 120 | 7.5 | 647, >750 | 120 | 89 (647 nm) 80 (750 nm) | RS˙− (750 nm) | Photocleavage reactions were anaerobic; sulfur spin adduct observed |
| 28 (ref. 71) | Binuclear dpq Cu(II) alkyl bridged complex | 669 | 90 | 7.5 | 647 | 120 | 13 (647 nm) | n.r. | Photocleavage reactions were anaerobic |
| 29 (ref. 72) | Tris(pyrazolyl) borate Cu(II) pyridyl-nap complex | 669 | 50 | 150 | >750 | 120 | 60 | ˙OH | Low chemical nuclease activity, dark cytotoxicity |
| 30 (ref. 73) | ONO donor Schiff base OV(IV) dppz complex | 694 | 40 | 67 | 753–799 | 120 | 28 | 1O2 | Low chemical nuclease activity |
| 31 (ref. 74) | Bis(dppz) OV(IV) complex | 715 | 32 | 10 | ≥750 | 120 | 61 | 1O2, ˙OH (365 nm) | Non-toxic in dark, highly photocytotoxic |
| ˙OH (750 nm) | |||||||||
| 32 (ref. 57) | Phenylated terpyridinyl OV(IV) imidazophen-pyrenyl complex | 745 | 65 | 30 | 705 | 120 | 68 | ˙OH | No dark cytotoxicity, higher phototoxicity in normal vs. cancer cells |
| 33 (ref. 57) | Glycosylated terpyridinyl OV(IV) imidazophen-pyrenyl complex | 684 | 96 | 30 | 705 | 120 | 72 | ˙OH | No dark cytotoxicity, higher phototoxicity in cancer vs. normal cells |
| 34 (ref. 46) | Pyridyl-phen OV(IV) curcumin complex | 757 | 76 | 50 | 785 | 60 | 48 | ˙OH | Low dark cytotoxicity, markedly higher phototoxicity in cancer vs. normal cells |
| 35 (ref. 44) | TPP-ph-terpyridine OV(IV) curcumin complex | 692 | 190 | 50 | 705 | 83 | ˙OH | High dark cytotoxicity; selective mitochondrial localization | |
| 36 (ref. 56) | Anthracenyl-benzimidazole OV(IV) dppz-Gly-Gly-OMe complex | 672 | 90 | 50 | 705 | 120 | 59 | ˙OH | Low dark cytotoxicity, high photocytotoxicity; selective mitochondrial localization |
| 37 (ref. 43) | Anthracenyl-bis(pyridyl)Fe(III) catecholate complex | 805 | 2400 | 25 | 785 | 60 | 83 | ˙OH | Low dark cytotoxicity, high photocytotoxicity; selective nuclear localization |
| 38 (ref. 82) | Bis(phenothiazinium) ethylenedipiperidine dye | 675 | 145000 |
5 | 710 | 60 | 93 | 1O2 > ˙OH | High photocleavage; cytotoxicity n.r. |
| 39 (ref. 82) | Mono(phenothiazinium) dye methylene blue | 661 | 78400 |
10 | 710 | 60 | 71 | 1O2 > ˙OH | Phototoxic in cancer cell lines16,110–112 |
| 40 (ref. 83) | H substituted N-methyl benz[e]indolium cyanine | 673 | 74000 |
10 | 700 | 60 | 18 | ˙OH > 1O2 (575 nm) | — |
| 41 (ref. 83) | Cl substituted N-methyl benz[e]indolium cyanine | 672 | 86000 |
10 | 700 | 60 | 33 | ˙OH > 1O2 (575 nm) | DNA affinity 41 > 40 |
| 42 (ref. 83) | Br substituted N-methyl benz[e]indolium cyanine | 669 | 83000 |
10 | 700 | 60 | 49 | ˙OH > 1O2 (575 nm) | DNA affinity 42 > 40 |
| 43 (ref. 83) | H substituted N-TMAB benz[e]indolium cyanine | 676 | 117000 |
10 | 700 | 60 | 34 | ˙OH > 1O2 (575 nm) | — |
| 44 (ref. 83) | Cl substituted N-TMAB benz[e]indolium cyanine | 680 | 134000 |
10 | 700 | 60 | 67 | ˙OH > 1O2 (575 nm) | Possible heavy atom effect vs. 43 |
| 45 (ref. 83) | Br substituted N-TMAB benz[e]indolium cyanine | 677 | 103000 |
10 | 700 | 60 | 59 | ˙OH > 1O2 (575 nm) | Possible heavy atom effect vs. 43 |
| 46 (ref. 84) | Bis(methylpyridinium) carbazole dye | 418 | 45500 |
30 | 800 | 35 | 100 | R3N˙+ | 2PA; equivalent aerobic, anaerobic cleavage yields |
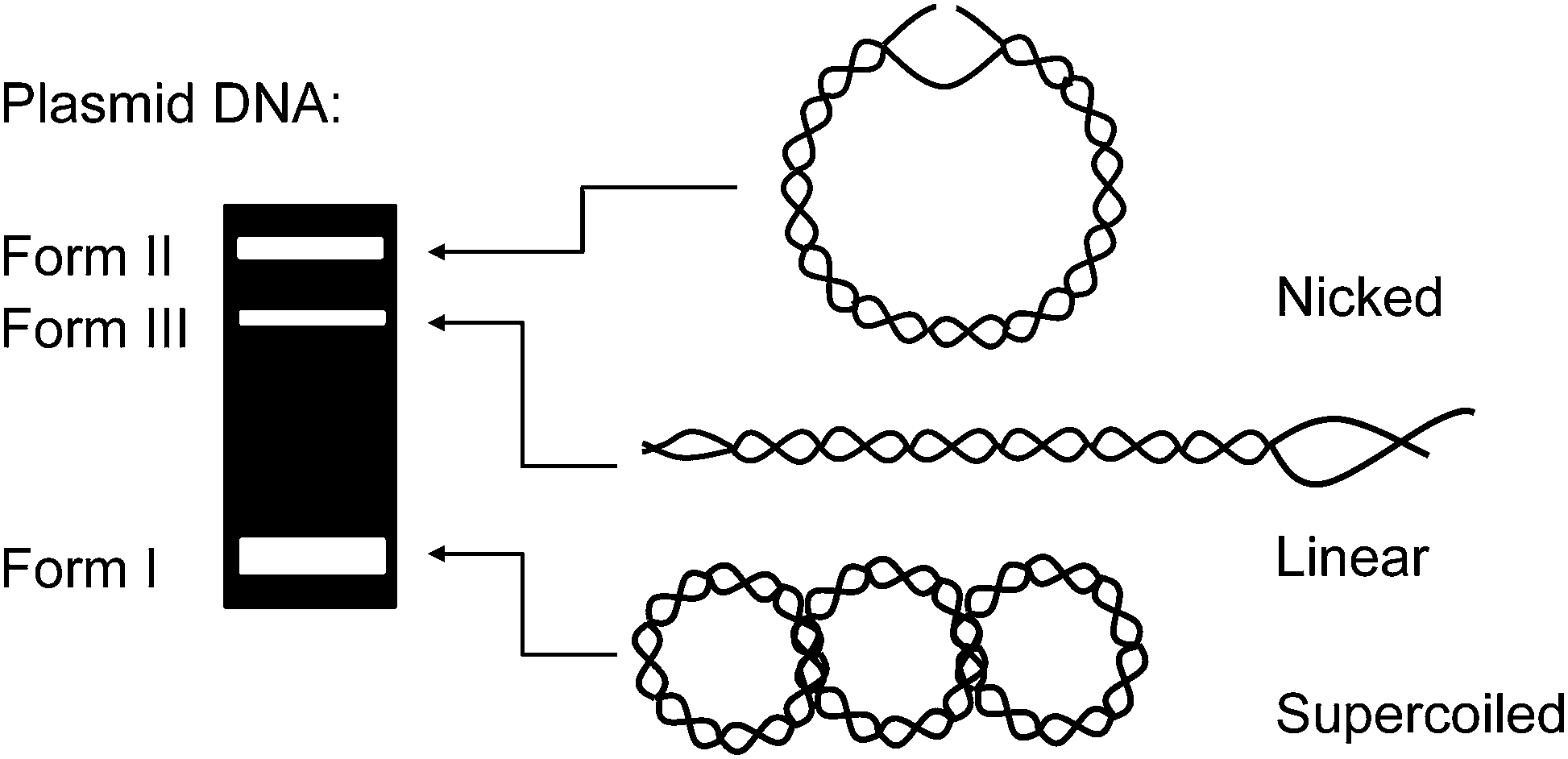 | ||
| Fig. 3 Plasmid conformations of equivalent molecular weight have dramatically different mobilities when electrophoresed through an agarose gel. In the DNA nicking assay shown above, the conversion of uncleaved supercoiled DNA (form 1) into nicked (form 2) and linear (form 3) DNA can be easily visualized after staining the gel with the fluorophore ethidium bromide. | ||
In contrast to lutetium(III) texaphyrin 20 (εmax at 732 nm ∼42000 M−1 cm−1), the low energy absorption maxima of most clinically available PDT agents are under 700 nm: e.g., porfimer sodium (12; εmax at 630 nm ∼3000 M−1 cm−1), verteporfin (13; εmax at 689 nm ∼34000 M−1 cm−1), temoporfin (17; εmax at 652 nm ∼35000 M−1 cm−1), and talaporfin (18; εmax at 664 nm ∼40000 M−1 cm−1).16 In order to effect a greater depth of light penetration, the possibility of utilizing lutetium(III) texaphyrin as a near-infrared PDT photosensitizing agent was therefore explored. Using a deeply penetrating 730 nm light source, a formulation based on lutetium(III) texaphyrin 22 (motexafin lutetium, Lutex®) was evaluated in phase I/II clinical trials for treatment of age related macular degeneration, breast cancer, cervical neoplasia, and prostate cancer.14,16 The rate of complete remission was 27% in a Phase I study in which lutetium(III) texaphyrin was administered to patients with locally recurrent breast cancer. Minimal toxicity to health surrounding tissue was observed.88 In a Phase I clinical trial involving patients with locally recurrent prostate cancer, PDT treatment with 22 induced a large transient increase in PSA levels.89 While treatment was generally well tolerated by patients, three clinical trials were terminated before the final study results were published.90
3. Copper(II) complexes
Paramagnetic transition metal coordination compounds possess low energy d–d absorption bands that, depending on the metal ion center and the ligand(s), can be extended into the far-red to near-infrared wavelength range. As a result, complexes containing different paramagnetic metal ion centers have been investigated for their DNA photocleaving properties.58–61,63,64 In earlier literature, there was considerable interest focused on the development of DNA photosensitizing agents based on the d9 metal ion copper(II). As a bio-essential element, copper(II) was initially expected to assist in ROS generation but to show low dark toxicity compared to heavier metals. However, increasing evidence emerged suggesting that Cu(II) complexes might not be ideal PDT agents. It was found that Cu(II) could be reduced to Cu(I) in the presence of biogenic reducing agents such as glutathione, NADPH and L-ascorbate,91–93 resulting in considerable dark toxicity in tissue culture experiments.43,72,73,75,79,80 Nonetheless, in this review we feature several important copper(II) complexes that give valuable insights into the photophysical mechanisms that facilitate low energy DNA photocleavage.The first copper(II) compounds to cleave DNA at excitation wavelengths ≥690 nm were polypyridyl complexes reported by Chakravarty and co-workers (Scheme 4, Table 1).67,68 In these 2003 experiments, a 694 nm pulsed ruby laser was used to target d–d absorption bands of Cu(II) bis-dipyridoquinoxaline (dpq) complex 23 and Cu(II) Schiff base-phenanthroline (phen) complex 24. After 120 and 30 min of irradiation, supercoiled plasmid was converted to nicked DNA in 98% and 50% yields in the presence of 50 μM of complexes 23 and 24, respectively. Chakravarty's photocleavage results were impressive taking into consideration the weak absorption exhibited by 23 (εmax at 673 nm ∼70 M−1 cm−1) and 24 (εmax at 662 nm ∼120 M−1 cm−1) in the far-red wavelength region. The molar extinction coefficients associated with the low energy absorption maxima of the DNA photocleaving agents lutetium(III) texaphyrin 20, TMPyPH2 21, and clinically relevant PDT agents such as porfimer sodium (12 in Scheme 2) and verteporfin (13) are considerably higher (3000 M−1 cm−1 to 42000 M−1 cm−1).
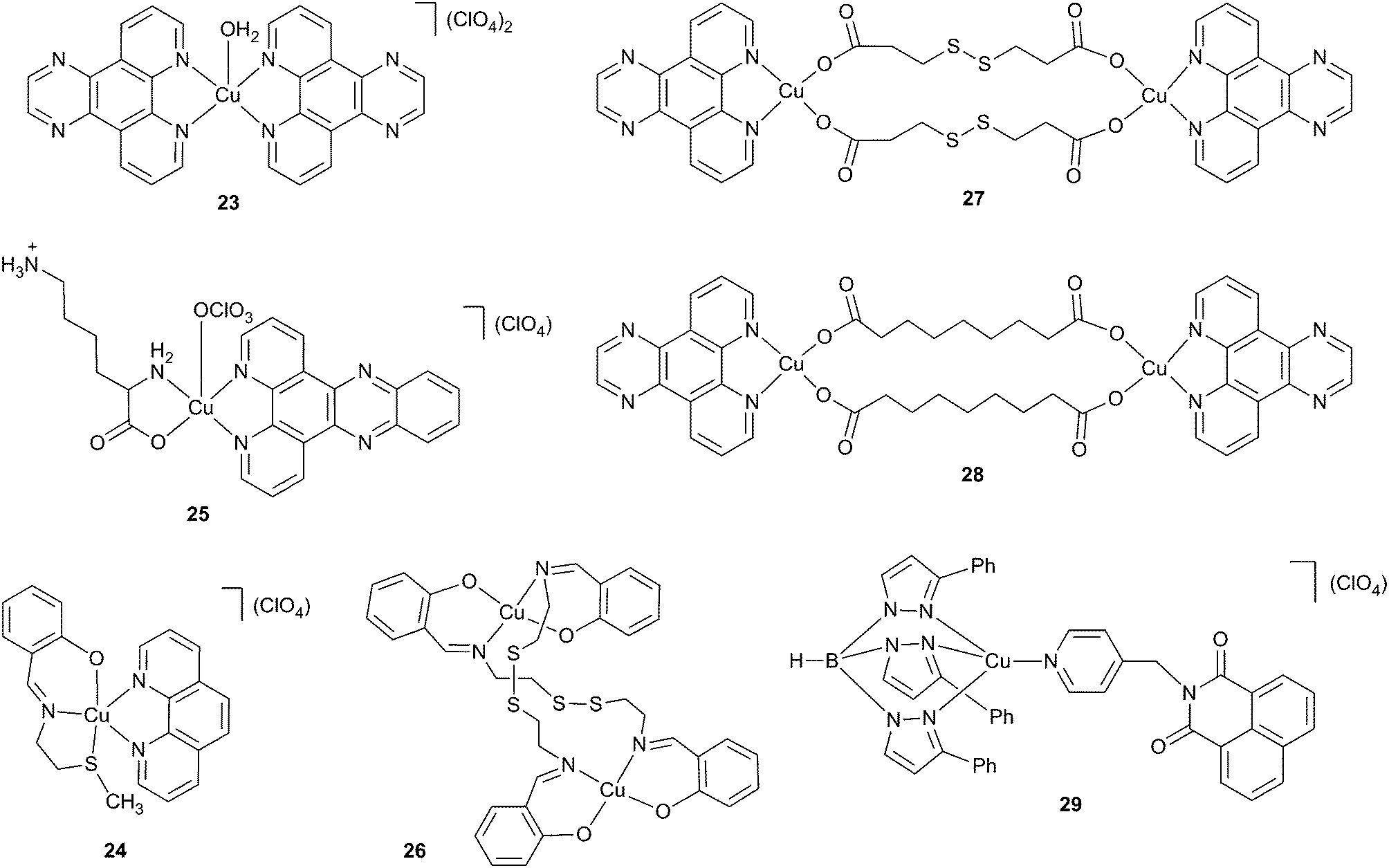 | ||
| Scheme 4 Long wavelength photonucleases based on copper(II). | ||
The dpq ligands of complex 23 were intended to serve dual roles as DNA binding elements and photosensitizing agents. Plasmid nicking assays conducted under argon or in the presence of ROS scavengers revealed DNA photocleavage involving two separate mechanisms.67 High energy 312 nm irradiation overlapping with charge transfer absorption sensitized the formation of Type II singlet oxygen, while low energy 632.8 nm irradiation gave rise to the production of Type I hydroxyl radicals. Although Cu(I) was not detected directly, Chakravarty and co-workers proposed that the reduction potential of the Cu(II)/Cu(I) couple might enable Cu(II) to undergo a one-electron reduction to Cu(I) via an electron transfer reaction in the excited state of 23. The reaction of ground state triplet oxygen with Cu(I) might then form hydroxyl radicals as shown in series of reactions in Scheme 5.91–94
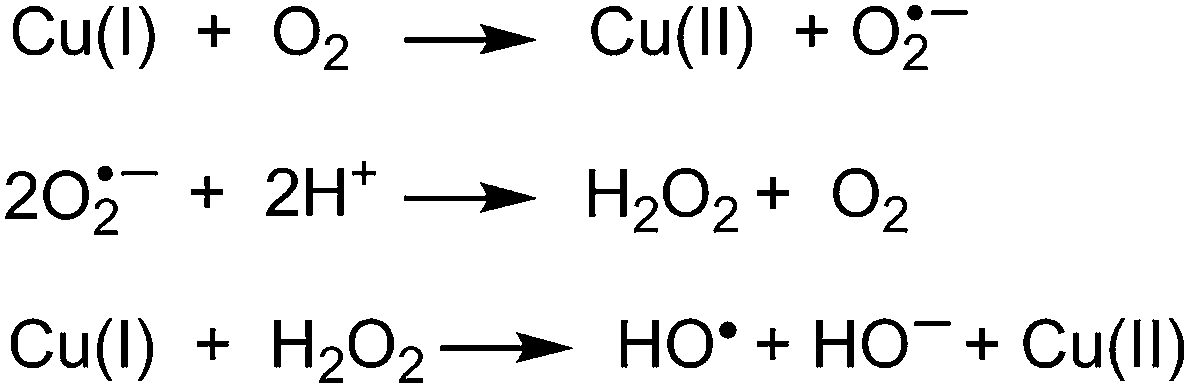 | ||
| Scheme 5 The reaction of HO− state triplet oxygen with Cu(I) generating the final product hydroxyl radicals. | ||
The phen ligand of complex 24 was employed to facilitate interactions within the DNA minor groove while the intended purpose of the tridentate NSO-donor Schiff base was to promote efficient intersystem crossing to the chromophore's excited triplet state.68 When the phen ligand was replaced by sterically imposing 2,9-dimethyl phenanthroline (dmp), DNA binding affinity accordingly decreased and the complex became photonuclease inactive. Photonuclease activity was also lost when the –SCH3 group of 24 was replaced by –OH. There was no cleavage when complex 24 was reacted with DNA in complete darkness. However, upon addition of the reducing agent 3-mercaptopropionic acid (MPA, 5 mM), there was significant, undesired chemical nuclease activity (no hν), highly suggestive of the potential for dark toxicity in tissue culture experiments. Pre-equilibrating DNA with distamycin reduced photocleavage yields, implying that complex 24 binds within the DNA minor groove. DNA photocleavage experiments conducted under argon and in the presence of ROS scavenging chemicals showed that irradiation of complex 24 at 532 nm and at 603 nm sensitizes the production of DNA damaging singlet oxygen.
Ray and co-workers utilized a polypyridyl copper(II) complex of L-lysine and dipyridophenazine (dppz) to cleave DNA upon irradiation with a Nd:YAG laser (25 in Scheme 4, Table 1).70 At 705, 720, 735, and 755 nm excitation wavelengths overlapping with d–d absorption, supercoiled DNA was converted into nicked plasmid in yields ranging from ∼67% to ∼97%. Evidence was presented for the involvement of Type II singlet oxygen in the cleavage reactions. Yields of nicked plasmid were reduced in the presence of the singlet oxygen scavenger sodium azide and enhanced by D2O, a solvent that increases the lifetime of singlet oxygen. Upon continuous irradiation of the complex, characteristic 1270 nm luminescence of 1O2 was detected. Nonetheless, it was not entirely clear that the reported cleavage was due to light activation. MPA (5 mM) was present in all of the reactions, the reducing agent shown to promote dark chemical nuclease activity in the case of complex 24 and other polypyridyl copper(II) compounds.61,68
In 2005, Chakravarty's group synthesized a square planar binuclear disulfide copper(II) complex in which each metal ion was bonded by two out of the four donors sites of a tetradentate, Schiff base bridging ligand ([Cu2(RSSR)2], R = 2-(thioethyl)salicylaldimine; 26 in Scheme 4, Table 1).69 Near complete cleavage of plasmid DNA occurred upon irradiating high concentrations of the complex with a 694 nm pulsed ruby laser (4 h, 400 μM of 26) or a 312 nm fluorescent trans illuminator (10 min, 200 μM of 26). Importantly, no dark chemical nuclease activity was detected when the complex was reacted with the reducing agents MPA and dithiothreitol (DTT). Similar to complex 23, DNA cleavage experiments conducted in the presence of ROS scavenging agents and D2O showed that 312 nm excitation of the charge transfer band of 26 promoted singlet oxygen production while 632.8 nm excitation of the complex's d–d band triggered hydroxyl radical formation (εmax at 306 nm = 7400 M−1 cm−1; εmax at 640 nm = 300 M−1 cm−1). Interestingly, Chakravarty and co-workers noted that 25% of plasmid DNA was cleaved upon 632.8 nm excitation under argon. This observation led the authors to hypothesize that two photocleavage mechanisms were likely to be operative. While aerobic light exposure at 632.8 nm generated ˙OH, anaerobic conditions might favor the formation of sulfide anion radicals capable of causing direct strand breaks in the plasmid DNA.
In a 2009 follow-up study, Chakravarty's group designed a series of binuclear copper(II) complexes in an attempt to find direct evidence for the involvement of sulfur-based radicals in anaerobic DNA cleavage.71 Two lead complexes contained dpq rings to promote DNA interactions and either the disulfide 3,3′-dithiodipropionic acid (dtdp; 27 in Scheme 4, Table 1) or nonanedioic acid (az; 28) as bridging ligands. A continuous wave Ar–Kr ion laser was used to irradiate the d–d bands of 27 and 28 for over 2 h (εmax of 27 at 661 nm = 120 M−1 cm−1; εmax of 28 at 669 nm = 90 M−1 cm−1). When 7.5 μM of dpq dtdp complex 27 was anaerobically irradiated with near-infrared light >750 nm, plasmid DNA was cleaved in 80% to 86% yield. In the presence of air, excitation of 27 and 28 at 647.1 nm generated DNA photocleavage in respective yields of 95% and 67% (pH 7.2). DNA viscosity and thermal melting measurements ruled against intercalation as a DNA binding mode, while pre-equilibration of plasmid DNA with methyl green suggested that complexes 27 and 28 interact with DNA in the major groove. When the 647.1 nm reactions were run under argon, photocleavage levels were 89% in the case of dtdp complex 27, but only 13% for az complex 28. This suggested that the sulfur-based dtdp bridging ligand was indeed instrumental in facilitating anaerobic DNA photocleavage. Inhibition of photocleavage in the presence of ROS scavenging agents identified hydroxyl radicals and singlet oxygen as sources of aerobic photonuclease activity. Cleavage levels were not reduced by the agents under anaerobic conditions, lending support to Chakravarty and co-workers' hypothesis that photolysis of the disulfide bond of 27 might form DNA damaging sulfide anion radicals. Several additional lines of evidence point to sulfur-based radical involvement. Although thiyl radicals (RS˙) are significantly less reactive than hydroxyl radicals, they are known to abstract hydrogen atoms from deoxyribose and are therefore theoretically capable of cleaving DNA.95 In experiments by Jain and coworkers, anaerobic DNA strand cleavage produced upon the reaction of glutathione (GSH) with Cu(II) was thought to involve the production of glutathione thiyl radicals (GS˙).96 When the phen analog of complex 27 was anaerobically irradiated with >750 nm light, Chakravarty and co-workers observed a 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) spin adduct with an EPR signal consistent with sulfide anion radical (RS˙−) or polysulfide radical (RS2−) formation.71,97 Type I abstraction of hydrogen atoms from DNA by a sulfur-based radical may indeed account for the photonuclease activity exhibited by complex 27 under anaerobic conditions.
As mentioned, it is theoretically possible for photosensitizing agents based on copper(II) to generate significant dark toxicity within cells by interacting with biogenic reducing agents such as glutathione. One electron reduction of the metal ion center followed by transfer of the electron to ground state triplet oxygen can trigger hydroxyl radical production (Scheme 5). With the aim of designing photosensitizers with greatly reduced dark toxicity, Chakravarty and co-workers synthesized a series of 3d-metal scorpionate complexes based on the formula [M(TpPh)(py-nap)](ClO4). The sterically imposing tridentate ligand tris(3-phenylpyrazolyl) borate (TpPh) was used to bind to and encapsulate a central divalent metal ion into a bowl-like structure, thus preventing its interaction with external reducing agents. The fourth coordination position of each complex was occupied by a pyridyl ligand modified with the DNA sensing chromophore 1,8-naphthalimide (py-nap). Cobalt(II), copper(II), and zinc(II) scorpionates exhibited enhanced emission upon intercalative binding of the naphthalimide unit to DNA. Importantly, low chemical nuclease activity was observed when the complexes were reacted in the presence of the reducing agent MPA. Only [Cu(II)(TpPh)(py-nap)](ClO4) (29) possessed a d–d absorption band in the far-red range (εmax at 669 nm = 50 M−1 cm−1). When high concentrations of this complex (150 μM) were irradiated at wavelengths >750 nm, plasmid DNA was cleaved in 60% yield (2 h). Radical scavenger and D2O experiments pointed to the involvement of hydroxyl radicals in the photocleavage reaction. The promising chemical nuclease and photocleavage results prompted testing of complex 29 in human cervical cell line (HeLa) tissue culture cells. Upon UV-A exposure, moderate photocytotoxicity was indeed observed (IC50 = 18.6 μM). Unfortunately, the compound was also toxic in the dark (IC50 = 29.7 μM), suggesting that intracellular redox processes were successful in reducing Cu(II) to Cu(I) by circumventing the sterically protective effects of the TpPh ligand.
4. Vanadium(IV) complexes
Of the metalated long-wavelength photonucleases reported in the literature, coordination compounds based on oxovanadium(IV) are by far the most well-studied.42,44,46,56,57,73–81 There are several advantages that make OV(IV) complexes potentially excellent PDT agents. While the d–d absorption bands of OV(IV) complexes are generally weaker in intensity, they extend further into the far-red to near-infrared wavelength range in comparison to copper(II) compounds.73 A second benefit is that the chemical nuclease activity of oxovanadium(IV)-based photonucleases is low (no hν). While the standard reduction potentials (E°) for OV(IV)/V(III) and Cu(II)/Cu(I) redox couples in acidic aqueous solutions are +0.337 V and +0.161 V respectively,98 under physiological conditions of temperature and pH, oxovanadium(IV) is not easily reduced to vanadium(III) by glutathione, NADPH, L-ascorbate and other common biogenic reducing agents.99,100 This avoids ROS production and the associated dark cytotoxicity that is expected in the case of many copper(II)-based photonucleases. In this section of the review, we have covered long-wavelength oxovanadium(IV) photonucleases considered to be representative of those published to date, with emphasis on compounds with novel chemical, physical, and biological properties.The first oxovanadium(IV) coordination compounds to cleave DNA at excitation wavelengths ≥690 nm were reported by Chakravarty and co-workers in 2007.73 These experiments focused on studying the DNA interactions of a series of photo-active OV(IV) complexes with O,N,O-donor α-amino acid Schiff base and N,N-donor polypyridyl ligands, either phen, dpq, or dppz. As expected, the compounds exhibited low chemical nuclease activity when treated with the reducing agent MPA (no hν). The best performing photonuclease in the series was [VO(salmet)(dppz)], where salmet = N-salicylidene-L-methionate and dppz = dipyridophenazine (30 in Scheme 6, Table 1; εmax at 694 nm ∼40 M−1 cm−1; εsh at 840 nm ∼20 M−1 cm−1). When 67 μM of 30 was irradiated from 753 nm to 799 nm for 2 h, plasmid DNA was photocleaved in 28% yield. Electrostatic and groove interactions were indicated by DNA thermal melting and viscometric titration data. Interestingly, D2O and ROS scavenger experiment with the analogous complex [VO(salmet)(dpq)] pointed to the exclusive involvement of singlet oxygen in the 753 nm to 799 nm photocleavage reaction, irradiation wavelengths that approach the theoretical 810 nm upper limit for 1O2 production.
 | ||
| Scheme 6 Long wavelength photonucleases based on oxovanadium(IV). | ||
In a 2009 follow-up study, Chakravarty and co-workers markedly improved DNA photocleavage outcomes with bis(dppz)-oxovanadium(IV) complex 31 (εmax at 715 nm ∼32 M−1 cm−1).74 After a 2 h exposure to ≥750 nm near-IR light, plasmid DNA was cleaved in 61% yield using a considerably lower concentration of compound (10 μM). Partial intercalative interactions in the DNA major groove were indicated by binding mode analyses. Scavenger and D2O experiments at ≥750 nm showed that hydroxyl radicals were the only contributors to DNA photocleavage. Similar to copper(II) dpq complexes 23 and 27, an independent mechanism involving singlet oxygen formation was identified when the light source was switched from near-infrared to ultra-violet.74 Bis(dppz)-oxovanadium(IV) complex 31 was then tested in human cervical cancer HeLa cells. The compound was found to be completely non-toxic in the dark, but was highly phototoxic upon exposure to 400 nm to 700 nm visible light (IC50 = 12.0 μM).
In addition to spatial targeting afforded by light activation, it would highly desirable for drugs in photodynamic cancer therapy to have the capacity to be selectively delivered to diseased cells in preference to normal cells. Cancer cells are known to over express glucose transporter proteins (GLUTs) on their surfaces and have significantly high rates of glucose uptake.101,102 One strategy to afford selective delivery would therefore be to attach a glucose moiety onto a PDT photosensitizing agent. Towards this end, Chakravarty and co-workers designed a series of oxovanadium(IV) complexes having either a phenylated or a glycosylated tridentate terpyridine ligand in addition to an imidazophenanthroline ligand with either an anthracenyl or pyrenyl fluorophore as a DNA sensing unit.57 No chemical nuclease activity was observed when the complexes were reacted with reducing agents MPA and glutathione (GSH). Optimal photocleavage results were then obtained with the phenylated (32 in Scheme 6, Table 1; εmax at 745 nm ∼65 M−1 cm−1) and glycosylated (33; εmax at 684 nm ∼96 M−1 cm−1) pyrenyl fluorophores. When reactions containing 30 μM of either 32 or 33 were irradiated at 705 nm for 2 h, plasmid DNA was cleaved in 68% to 72% yields, respectively. Scavenger and D2O experiments at 705 nm pointed to the involvement of hydroxyl radicals in the photocleavage reaction, while significant intercalative DNA binding was indicated by thermal melting and viscometric titration assays. The hypothesis of cell-specific drug localization was then supported by time-dependent monitoring of the emission of 32 and 33 in a fluorescence activated cell sorting (FACS) experiment. In other words, glycosylated 33 was incorporated by cancer cells about 3-fold faster as compared to its phenylated analogue 32. Both 32 and 33 were non-toxic in the dark (IC50 > 100 μmol). Upon irradiation with a 400–700 nm light source, glycosylated complex 33 was more phototoxic in HeLa cancer (IC50 = 1.9 ± 1.1 μmol) and Hep G2 cancer (IC50 = 2.4 ± 1.0 μmol) cells over normal HEK 293T cells (IC50 = 5.5 ± 0.9 μmol). A comparison of IC50 values shows that replacing the phenyl unit of 32 with glycose in 33 increases phototoxicity in cancer cells, while having essentially no effect on phototoxicity levels in normal cells. In sum, the research study suggests that it should be possible to design oxovanadium(IV) PDT agents that can not only be spatially targeted by light but can be delivered selectively to specific cell types.57
In a subsequent publication, Chakravarty and co-workers described the synthesis and DNA interactions of additional photo-active oxovanadium(IV) nucleases capable of displaying cell-selective phototoxicity. The study's lead complex [VO(pyphen)(cur)](ClO4) consisted of curcumin (cur) and a DNA binding 2-(2′-pyridyl)-1,10-phenanthroline (pyphen) ligand (34 in Scheme 6, Table 1; εmax at 757 nm ∼76 M−1 cm−1; εsh at 805 nm ∼68 M−1 cm−1).46 Curcumin, a natural product extracted from Curcuma longa Linn (turmeric), was selected based on its known anti-cancer activity and ability to generate ROS upon light activation.103,104 When irradiated at 785 nm, oxovanadium(IV) complex 34 cleaved plasmid DNA in 48% yield. Under 400–700 nm illumination, the curcumin complex was found to be significantly more phototoxic in HeLa cancer (IC50 = 3.4 ± 0.3 μM) and MCF-7 cancer (IC50 = 4.5 ± 0.2 μM) cells compared to normal 3T3 cells (IC50 = 20.6 ± 0.6 μmol). Dark toxicity was low in all cases (IC50 > 50 μM). Cancer cell-selective photocytotoxicity was also observed when using [VO(pyphen)(anacac)](ClO4), a complex in which the curcumin unit of 34 was replaced by anthracenylacetylacetonate (anacac). This suggested that the pyphen ligand, in common to the curcumin and anacac complexes, might have potentially played a role in cancer cell selectivity. Mechanistic studies indicated that hydroxyl radicals contributed to photocleavage and that DNA binding was partially intercalative in nature. Because curcumin complex 34 emits green fluorescence, its cellular uptake and localization could be directly monitored in HeLa and MCF-7 cells. The compound was found to localize in the cell cytoplasm in addition to the nucleus, pointing to genomic DNA as a potential sub-cellular target. In so doing, the study demonstrated that compound 34 might have the potential to play dual roles as a tumor selective therapeutic agent in photodynamic therapy and as a fluorescent probe in tumor detection and imaging.
Anti-cancer agents that trigger apoptosis are of great interest in drug design due to the propensity of highly proliferative cancer cells to evade programmed cell death.105 Moreover, PDT agents that generate ROS have been shown to induce an apoptotic response after photo-damage to mitochondria, but not to other organelles.106,107 Photosensitizers that efficiently localize in the mitochondria are therefore highly sought after in PDT. Towards this end, Chakravarty and co-workers synthesized oxovanadium(IV) curcumin complex [VO(TPP-ph-tpy)(cur)](ClO4) having TPP-ph-tpy, a terpyridyl ligand (typ) modified with the mitochondrial targeting p-triphenylphosphonium methylphenyl bromide TPP-ph group (35 in Scheme 6, Table 1; εmax at 692 nm ∼190 M−1 cm−1).44 Under 705 nm illumination, a 50 μM concentration of complex 35 was shown to cleave plasmid DNA by a hydroxyl radical pathway (83% yield). Because curcumin is fluorescent, uptake into HeLa cells and sub-cellular localization were then visualized by microscopy. The analysis showed that TPP-ph-tpy complex 35 localized exclusively inside mitochondria, pointing to mitochondrial DNA as a possible drug target. In contrast, the control complex [VO(ph-tpy)(cur)](ClO4), in which the TPP-ph unit of 35 was replaced by a simple phenyl ring (ph), showed no localizational preference and was found inside a number of organelles. When tested in HeLa and MCF-7 cancer cells, TPP-ph-tpy complex 35 unfortunately displayed significant dark toxicity (IC50 = 17.0 μM to 22.4 μM) compared to the ph-tpy dark control (IC50 > 100 μM). This suggested that TPP-ph was conferring the toxicity onto complex 35, placing possible limits on the therapeutic potential of the triphenylphosphonium unit as a mitochondrial targeting group.44
In a related study, Chakravarty and co-workers achieved low dark toxicity, high photocytotoxicity, and specific mitochondrial targeting with long wavelength oxovanadium(IV) photonuclease 36 (εmax at 672 nm ∼90 M−1 cm−1). The complex consisted of a benzimidazole ligand appended with an anthracenyl fluorophore and dppz conjugated to the dipeptide Gly-Gly-OMe.56 When reactions containing 50 μM of 36 were irradiated at 705 nm for 2 h, plasmid DNA was cleaved in 59% yield. Mechanistic experiments showed that the photocleavage reaction occurred by a hydroxyl radical pathway and that DNA binding was likely to involve partial intercalation. Sub-cellular localization of complex 36 was then monitored by fluorescence microscopy. A merged microscopic image of blue anthracene fluorescence with a second image of mitotracker deep red showed that complex 36 exclusively localized in the mitochondria of HeLa and HaCaT cancer cells. Using the mitochondrial dye Rhodamine 123 in FACS experiments, disruption of the mitochondrial membranes by 36 was observed upon irradiation with 400–700 nm light, a hallmark of apoptotic cell death. Apoptosis was then confirmed in a DNA laddering assay. Using the ROS specific fluorescence probe dichlorofluorescein in combination with mitotracker deep red, confocal microscopic images showed that irradiation of HeLa and HaCaT cells equilibrated with complex 36 triggered ROS production exclusively inside mitochondria. In complex 36 treated HeLa and HaCaT cells, significant 400–700 nm photocytotoxicity (IC50 = 9.5 ± 0.2 μM to 14.8 ± 0.9 μM) was observed compared to when the cells were kept in the dark (IC50 = 44.0 ± 0.1 μM to >50 μM) or to when they were photo-irradiated in the presence of the ROS quencher N-acetyl-L-cysteine (IC50 = 48.3 ± 0.5 μM to >50 μM). Taken together, multiple lines of evidence presented in the paper suggest that irradiation of 36 triggered ROS generation inside of the mitochondria of cancer cells, causing apoptosis to occur. This finding is noteworthy taking into consideration the strong tendency of many highly proliferative cancer cells to evade programmed cell death.105
5. Iron(III) complex
Similar to copper(II) and oxovanadium(IV), iron(III) is a redox active bio-essential transition metal ion that is of considerable importance for maintaining a healthy state.108 To avoid the weak molar extinction coefficients associated with the d–d absorption bands of long wavelength photonucleases based on copper(II) and oxovanadium(IV), Chakravarty and co-workers designed a ternary iron(III) complex that was capable of efficiently cleaving nucleic acids under 785 nm illumination (37 in Scheme 7, Table 1).43 The pendant anthracenyl moiety of 37 was incorporated to serve as a fluorophore as well as a DNA binding element, whereas a catecholate unit was employed to create an intense ligand-to-metal charge transfer (LMCT) absorption band within the near-infrared PDT window. Spectral analysis showed a visible band at 805 nm (εmax = 2400 M−1 cm−1) corresponding to the catecholate–Fe(III) LMCT transition. When 25 μM of complex 37 was irradiated at 785 nm for 60 min, DNA photocleavage occurred in high yield, with 83% of supercoiled plasmid being converted to the nicked form. ROS scavenger and D2O studies pointed to the involvement of hydroxyl radicals in the cleavage mechanism, while DNA thermal melting and viscosity measurements suggested that binding with DNA was partially intercalative in nature. Interestingly, the standard reduction potential for the Fe(III)/Fe(II) redox couple in acidic aqueous solution is high (+0.771 V).98 Moreover, under physiological conditions of temperature and pH, Fe(III) is easily reduced to Fe(II) by biogenic reducing agents such as GSH, NADPH, and L-ascorbate, leading to facile hydroxyl radical production by an H2O2-dependent Fenton reaction (e.g., final step in Scheme 5).109 In the case of complex 37 however, no chemical nuclease activity was observed upon H2O2 and GSH addition. Dark toxicity in HeLa cancer cell lines (IC50 > 100 μM) was significantly lower than for the widely used PDT agent porfimer sodium (IC50 > 41 μM, 12 in Scheme 2). On the other hand, complex 37 (IC50 = 6.2 ± 0.1 μM) and porfimer sodium (IC50 = 4.3 ± 0.2 μM) were both highly phototoxic when the HeLa cells were irradiated with a 400–700 nm light source.43 The complex was also phototoxic to the HeLa cells under 600–720 nm long wavelength illumination (IC50 = 18.8 ± 0.1 μM). Time dependent analysis of anthracene fluorescence by laser scanning confocal microscopy showed almost complete accumulation of 37 into the cell nucleus, underscoring the importance of genomic DNA as a potential subcellular target of the complex. | ||
| Scheme 7 Long wavelength photonuclease based on iron(III). | ||
6. Non-metalated photosensitizers
Conventional non-metalated DNA photosensitizers described in the literature have the advantage of possessing extended π systems that have the potential to achieve strong absorption of light in the far-red to near-infrared range. In 2008, Grant, Lorente, and co-workers described the synthesis of a strongly absorbing bis-phenothiazinium intercalating agent with two phenothiazine units covalently connected by a semi-rigid ethylenedipiperidine linker (38 in Scheme 8, Table 1; εmax (1% SDS) at 675 nm ∼145000 M−1 cm−1).82 The synthetic design of 38 was based on the well-known photosensitizer methylene blue (MB) (39 in Scheme 8, Table 1; εmax (1% SDS) at 661 nm ∼78400 M−1 cm−1), a mono-phenothiazinium dye that demonstrates efficient light-induced activity against human lung adenocarcinoma, bladder carcinoma, and melanoma cells in tissue culture experiments.16,110–112 Similar to strongly absorbing lutetium(III) texaphyrin 20 (εmax at 732 nm ∼42000 M−1 cm−1),16 DNA nicking assays showed that 38 could efficiently photocleave plasmid DNA at low micro molar concentrations. Upon irradiation at 710 nm for 1 h, the yield produced by 5 μM of bis-phenothiazine 38 (93%) was higher than the yield generated by 2 mol equiv. of the mono-phenothiazinium dye MB (71%). In both cases, scavenger and D2O experiments at 710 nm pointed to singlet oxygen in the major cleavage pathway with a minor contribution from hydroxyl radicals. As in the case of MB, polyacrylamide gel electrophoresis of compound 38 at nucleotide resolution showed a strong preference for strand breaks at guanine bases, a finding supporting singlet oxygen involvement. DNA viscosity and melting temperature measurements provided evidence substantiating the existence of three DNA binding modes for bis-phenothiazine 38. In addition to bis-intercalation, classical mono-intercalation and groove binding interactions were indicated.82
 | ||
| Scheme 8 Long wavelength non-metalated photonucleases. | ||
Cyanine dyes have found promising applications in medicine as fluorescent probes for biomolecular imaging and as photosensitizing agents in photodynamic therapy.53,113–117 The typical structure of a cyanine dye consists of two terminal positively charged heterocyclic ring systems containing nitrogen atoms connected by a central polymethine bridge. Common heterocycles include indole, quinoline, benzoxazole, and benzothiazole rings. Since the bridge has an odd number of carbons, the positive charge is delocalized over the two ring nitrogen atoms via conjugation. By lengthening the polymethine chain and by optimizing the heterocyclic units, the optical properties of cyanine dyes can be tuned to afford strong absorption of light in the far-red to near-infrared wavelength range.118 Indocyanine green (ICG) is an FDA-approved cyanine drug indicated for ophthalmic angiography, sentinel lymph node detection in cancer surgery, and cardiac output, hepatic function, and liver blood flow monitoring.114,115 Recent studies have demonstrated that ICG and other cyanine dyes produce considerable long wavelength-induced photocytotoxicity in breast cancer, squamous cell carcinoma, and melanoma cell lines.53,113,116
In 2014, Grant, Henary, and co-workers studied the DNA interactions of a series of six symmetrical benz[e]indolium carbocyanine dyes (40 to 45 in Scheme 8, Table 1).83 The terminal aromatic rings of each cyanine were intended to promote DNA interactions while a central pentamethine bridge was selected to achieve long wavelength light absorption. Halogen introduction at the meso position of the central bridge (Cl in 41 and 44, Br in 42 and 45 vs. H in 40 and 43) was intended to induce a “heavy atom effect”, known to promote intersystem crossing of photosensitizers from singlet to triplet excited states and thus enhance ROS formation.119 In the case of mono-cationic dyes 40 to 42, methyl groups were appended to the heterocyclic nitrogen atoms to minimize steric clashes with DNA, while (3-bromopropyl)trimethylammonium bromide (TMAB) groups bearing positive charge were employed in tri-cationic dyes 43 to 45 to optimize aqueous solubility and enhance electrostatic DNA interactions. UV-visible spectra showed intense long wavelength monomer and H-aggregate absorption bands. The TMAB-substituted compounds, e.g., 43 (εmax at 676 nm ∼117000 M−1 cm−1), 44 (εmax at 680 nm ∼134000 M−1 cm−1), and 45 (εmax at 677 nm ∼103000 M−1 cm−1) displayed stronger monomer absorption than the cyanines in the methyl-substituted series, e.g., 40 (εmax at 673 nm ∼74000 M−1 cm−1), 41 (εmax at 672 nm ∼86000 M−1 cm−1), and 42 (εmax at 669 nm ∼83000 M−1 cm−1). Plasmid nicking assays were conducted at a dye concentration of 10 μM using low power 700 nm LED lamps. Within the mono-cationic methyl and tricationic TMAB series, the halogenated carbocyanines produced more photocleavage than their non-halogenated counterparts. This suggested either that the halogenated dyes were binding to DNA with higher affinity or that their chlorine and bromine atoms were increasing DNA photocleavage by a heavy atom effect. After 60 min of irradiation, chlorinated TMAB cyanine 44 displayed the highest activity with a 67% plasmid DNA cleavage yield, followed by brominated TMAB 45 (59%), and brominated methyl cyanine 42 (49%). The involvement of hydroxyl radicals and singlet oxygen in the photocleavage reactions was supported by ROS scavenger experiments, while DNA-induced spectral changes in UV-visible absorption and fluorescence spectra pointed to non-intercalative dye–nucleic acids interactions.
The theory of two-photon absorption (2PA) was first developed by Maria Göppert-Mayer in the 1930s. In her doctoral dissertation, she predicted the existence of a process involving excitation of an atom to an excited state by two simultaneously absorbed photons. The phenomenon was experimentally demonstrated in 1961 shortly after the invention of the laser. Unlike one-photon absorption (1PA), which depends linearly on incident light intensity, two-photon absorption is proportional to the square of the irradiance, resulting in a great reliance on intense laser beams (e.g., picosecond and femtosecond pulsed lasers). Most importantly, the 2PA process enables excitation of a molecule by employing photons with half the energy (or twice the wavelength) of the corresponding 1PA transition. This property minimizes photochemical decomposition of the chromophore, reduces light scattering, and allows deeper penetration of the incident light through biological tissues. Therefore, there is a great interest in the development of efficient two-photon dyes for clinical use as fluorescent imaging probes and photodynamic therapy agents. Typical 2PA dyes consist of electron-donating (D) and/or electron-accepting (A) groups connected by π-conjugated bridges. Well-explored arrangements include D–π–D, A–π–A, D–π–A, and D–π–A–π–D systems. The 2PA cross-section (δ2PA) is a useful parameter for evaluating the efficiency of a 2PA chromophore and can be maximized by increasing the length and planarity of the bridge, e.g., by introducing acetylene units to ensure that the bridge cannot be twisted out of conjugation.120,121
In a 2015 RSC Advances communication, Zheng, Duan, and co-workers reported highly efficient, unprecedented two-photon DNA cleavage with near-infrared 800 nm light under anaerobic conditions.84 Chromophore 46 (Scheme 8, Table 1) was designed with a symmetric A–π–D–π–A structure consisting of a central carbazole unit as the electron donor connected to terminal methylpyridinium acceptors via acetylene. This afforded a two-photon DNA photosensitizing agent with a high 2PA cross-section (δ2PA at 800 nm = 401 GM, where 1 GM = 10−50 cm4 s per photon per molecule). The 418 nm charge transfer band of 46 (εmax ∼ 45500 M−1 cm−1) was intended to promote strong two-photon absorption of 800 nm light, while planar aromatic ring systems with positive charges were employed to enhance water solubility and DNA interactions. Upon the addition of CT DNA, compound 46 exhibited significant fluorescence enhancement and spectral changes in UV-visible absorption, indicating strong DNA–ligand binding by external-electrostatic and intercalative modes. DNA nicking assays were then carried out upon illumination with an 800 nm femtosecond pulsed laser for 35 min. Complete, 100% photo-conversion of plasmid DNA from the supercoiled to nicked form was accomplished at a concentration of 46 equal to 30 μM. Superoxide dismutase and D-mannitol had no apparent inhibitory effects on DNA cleavage, ruling against hydroxyl radical involvement. Equivalent photocleavage yields were achieved under air-saturated and nitrogen atmospheres, pointing to an anaerobic Type I DNA strand breakage mechanism involving electron transfer. EPR spin trapping of free radicals with DMPO identified the formation of N-centered amine radical cations, which were proposed to cause DNA cleavage via abstraction of hydrogen atoms from deoxyribose. Zheng and Duan's study is noteworthy for several reasons. Using two-photon chromophore 46, plasmid was cleaved in quantitative yields under 800 nm illumination, the longest wavelength of light successfully utilized in DNA photocleavage experiments published to date. Moreover, the results obtained show great promise that the PDT approach may one day be extended to include the treatment of solid tumors that thrive under hypoxic conditions.20 The authors report that photocytotoxicity and in vivo PDT experiments are currently in progress.
7. Concluding remarks
Photosensitizers that cleave DNA are important in the development of anti-cancer agents in photodynamic therapy. Excitation with low energy irradiation activates the PDT agents, causing highly localized, oxidative damage to DNA and other macromolecules in targeted cells. This allows artificial control over the location and duration of anti-cancer treatment, contributing to high selectivity and promise of the PDT method to reduce the severe adverse side effects associated with many conventional chemotherapeutic agents.While most routinely used PDT photosensitizers are activated by light sources that emit at wavelengths ≤689 nm, excitation that extends from the far-red into the near-infrared range is preferable because irradiation in this region has a greater penetration depth through biological tissue. In this review, we have described the development of novel metalated and non-metalated photosensitizing agents that cleave DNA when illuminated at wavelengths ≥690 nm. Among the metallo-photonucleases that have been described are coordination compounds based on Lu(III), Cu(II), V(IV), and Fe(III). Potential advantages of introducing metal ions into photosensitizing agents include their ability to generate d–d transition and/or charge transfer absorption bands that overlap with PDT excitation wavelengths in the far-red to near-infrared range. The undesired dark cellular toxicity of copper(II) complexes led to the development of oxovanadium(IV) and iron(III) coordination compounds that were toxic to cells only under illumination. The metal-based complexes and non-metalated chromophores both proved to be excellent long wavelength photonucleases, in several cases achieving highly efficient DNA cleavage in the presence of low micro molar concentrations of compound. While strong absorption of long wavelength light was achieved by the lutetium(III) texaphyrin and anthracenyl iron(III) catecholate complexes, the extended π systems of the non-metalated cyanine, phenothiazine, and two-photon carbazole chromophores were found to possess superior molar absorptivity compared to the weak d–d absorption bands associated with copper(II) and oxovanadium(IV) transition metal complexes. In the case of non-metalated agents, there is no need for concern that coordinated paramagnetic metal centers will shorten the lifetime of the excited triplet state of the photosensitizer or that one electron chemical reduction of metal ions will generate dark cytotoxicity.
Reaction pathways leading to DNA photocleavage by the metalated and non-metalated photosensitizing agents included aerobic Type I electron transfer and aerobic Type II energy transfer processes in addition to anaerobic Type I hydrogen atom abstraction from deoxyribose by sulfide anion and amine-based radicals. The most common mechanism encountered was an oxygen-dependent, Type I hydroxyl radical pathway, especially upon irradiation in the near-infrared wavelength region. Considering that there is no requirement for molecular oxygen, the anaerobic photonucleases show great potential as agents for hypoxic tumor treatment. The majority of the long wavelength photosensitizers possessed extended aromatic ring systems that served as DNA binding and/or sensing units. Appending the metalated photosensitizers with chemical tags was shown to facilitate their selective delivery to apoptosis-inducing mitochondria and to cancer cells. While tremendous progress has been made by scientists in the PDT field, caution is requested when one attempts to compare the photonucleases described in this review, as various light sources, reagent concentrations, irradiation times, and DNA preparations were employed in the different experiments we have described. After all, it is our hope that researchers will gain benefit from this summary in their future attempts to develop DNA photonucleases that are activated by far-red to near-infrared light.
References
- J. Y. Chen and J. Stubbe, Nat. Rev. Cancer, 2005, 5, 102–112 CrossRef CAS PubMed
.
- R. Palchaudhuri and P. J. Hergenrother, Curr. Opin. Biotechnol., 2007, 18, 497–503 CrossRef CAS PubMed
- R. Ralhan and J. Kaur, Expert Opin. Ther. Pat., 2007, 17, 1061–1075 CrossRef CAS
- T. C. Johnstone, G. Y. Park and S. J. Lippard, Anticancer Res., 2014, 34, 471–476 CAS
- V. Murray, J. K. Chen and A. M. Galea, Cell. Mol. Life Sci., 2014, 71, 1505–1512 CrossRef CAS PubMed
- C. Tang, A. Paul, M. P. Alam, B. Roy, W. D. Wilson and S. M. Hecht, J. Am. Chem. Soc., 2014, 136, 13715–13726 CrossRef CAS PubMed
- T. Taşkın-Tok and S. Gowder, Anticancer drug—friend or foe, InTech, Rijeka, 2014 Search PubMed
- P. Lu, Y. J. Li and D. Y. Hu, Curr. Org. Chem., 2015, 19, 958–968 CrossRef CAS
- Z. Yu, R. Paul, C. Bhattacharya, T. Bozeman, M. Rishel and S. Hecht, Biochemistry, 2015, 54, 3100–3109 CrossRef CAS PubMed
- H. Joensuu, Lancet Oncol., 2008, 9, 304 CrossRef PubMed
- Convention on the Prohibition of the Development, Production, Stockpiling and use of Chemical Weapons and their Destruction, http://www.opcw.nl, accessed October, 2015.
- A. P. Castano, T. N. Demidova and M. R. Hamblin, Photodiagn. Photodyn. Ther., 2004, 1, 279–293 CrossRef CAS PubMed
- A. Juzeniene and J. Moan, Photodiagn. Photodyn. Ther., 2007, 4, 3–11 CrossRef CAS PubMed
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed
- L. B. Josefsen and R. W. Boyle, Theranostics, 2012, 2, 916–966 CrossRef CAS PubMed
- A. B. Ormond and H. S. Freeman, Materials, 2013, 6, 817–840 CrossRef CAS
- I. Yoon, J. Z. Li and Y. K. Shim, Clinical Endoscopy, 2013, 46, 7–23 CrossRef PubMed
- R. Bonnett, Chem. Soc. Rev., 1995, 24, 19–33 RSC
- J. Moan and K. Berg, Photochem. Photobiol., 1991, 53, 549–553 CrossRef CAS PubMed
- S. Hatz, L. Poulsen and P. R. Ogilby, Photochem. Photobiol., 2008, 84, 1284–1290 CrossRef CAS PubMed
- G. M. Makrigiorgos, Met. Ions Biol. Syst., 1999, 36, 521–545 CAS
- G. Viola and F. Dall'Acqua, Curr. Drug Targets, 2006, 7, 1135–1154 CrossRef CAS PubMed
- C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233, 351–371 CrossRef
- M. Ochsner, J. Photochem. Photobiol., B, 1997, 39, 1–18 CrossRef CAS
- E. Clo, J. W. Snyder, P. R. Ogilby and K. V. Gothelf, ChemBioChem, 2007, 8, 475–481 CrossRef CAS PubMed
- K. König, Handbook of Biomedical Nonlinear Optical Microscopy, Oxford University Press, New York, 2008 Search PubMed
- S. K. Powers and M. R. Detty, in Photodynamic Therapy of Neoplastic Disease, ed. D. Kessel, CRC Press, Boca Raton, 1990, vol. 1, ch. 19, pp. 307–328 Search PubMed
- P. M. Wood, Biochem. J., 1988, 253, 287–289 CrossRef CAS PubMed
- B. F. Overholt, C. J. Lightdale, K. K. Wang, M. I. Canto, S. Burdick, R. C. Haggitt, M. P. Bronner, S. L. Taylor, M. G. A. Grace and M. Depot, International Photodynamic Group for High-Grade Dysplasia in Barrett's Esophagus, Gastrointest. Endosc., 2005, 62, 488–498 CrossRef PubMed
- T. Tanaka, S. Matono, T. Nagano, K. Murata, S. Sueyoshi, H. Yamana, K. Shirouzu and H. Fujita, Gastrointest. Endosc., 2011, 73, 1–6 CrossRef PubMed
- L. Corti, L. Toniolo, C. Boso, F. Colaut, D. Fiore, P. C. Muzzio, M. I. Koukourakis, R. Mazzarotto, M. Pignataro, L. Loreggian and G. Sotti, Lasers Surg. Med., 2007, 39, 394–402 CrossRef PubMed
- X. J. Cai, W. M. Li, L. Y. Zhang, X. W. Wang, R. C. Luo and L. B. Li, Photodiagn. Photodyn. Ther., 2013, 10, 672–676 CrossRef PubMed
- J. W. Miller, U. Schmidt-Erfurth, M. Sickenberg, C. J. Pournaras, H. Laqua, I. Barbazetto, L. Zografos, B. Piguet, G. Donati, A. M. Lane, R. Birngruber, H. van den Berg, A. Strong, U. Manjuris, T. Gray, M. Fsadni, N. M. Bressler and E. S. Gragoudas, Arch. Ophthalmol., 1999, 117, 1161–1173 CrossRef CAS PubMed
- P. K. Lee and A. Kloser, J. Drugs Dermatol., 2013, 12, 925–930 CAS
- S. A. de Visscher, P. U. Dijkstra, I. B. Tan, J. L. Roodenburg and M. J. Witjes, Oral Oncol., 2013, 49, 192–210 CrossRef CAS PubMed
- J. Usuda, H. Kato, T. Okunaka, K. Furukawa, H. Tsutsui, K. Yamada, Y. Suga, H. Honda, Y. Nagatsuka, T. Ohira, M. Tsuboi and T. Hirano, J. Thorac. Oncol., 2006, 1, 489–493 CrossRef PubMed
- Y. P. Istomin, M. A. Kaplan, S. V. Shliakhtsin, T. P. Lapzevich, D. A. Cercovsky, L. N. Marchanka, A. S. Fedulov and T. V. Trukhachova, Proc. SPIE, 2009, 7380, 73806V CrossRef
- E. W. Cerrati, S. A. Nguyen, J. D. Farrar and E. J. Lentsch, Ear Nose Throat. J., 2015, 94, 72–79 Search PubMed
- B. C. Wilson, in Photodynamic Therapy, ed. T. Patrice, Royal Society of Chemistry, Cambridge, 2003, ch. 7 pp. 127–158 Search PubMed
- S. Stolik, J. A. Delgado, A. Perez and L. Anasagasti, J. Photochem. Photobiol., B, 2000, 57, 90–93 CrossRef CAS
- B. Banik, P. K. Sasmal, S. Roy, R. Majumdar, R. R. Dighe and A. R. Chakravarty, Eur. J. Inorg. Chem., 2011, 1425–1435 CrossRef CAS
- U. Basu, I. Khan, A. Hussain, P. Kondaiah and A. R. Chakravarty, Angew. Chem., Int. Ed. Engl., 2012, 51, 2658–2661 CrossRef CAS PubMed
- B. Banik, K. Somyajit, G. Nagaraju and A. R. Chakravarty, Dalton Trans., 2014, 43, 13358–13369 RSC
- C. Mari, V. Pierroz, R. Rubbiani, M. Patra, J. Hess, B. Spingler, L. Oehninger, J. Schur, I. Ott, L. Salassa, S. Ferrari and G. Gasser, Chem.–Eur. J., 2014, 20, 14421–14436 CrossRef CAS PubMed
- S. Banerjee, A. Dixit, A. A. Karande and A. R. Chakravarty, Eur. J. Inorg. Chem., 2015, 447–457 CrossRef CAS
- S. Rangasamy, H. Ju, S. Um, D. C. Oh and J. M. Song, J. Med. Chem., 2015, 58, 6864–6874 CrossRef CAS PubMed
- M. B. Vrouenraets, G. W. Visser, G. B. Snow and G. A. van Dongen, Anticancer Res., 2003, 23, 505–522 CAS
- J. A. Woods, N. J. Traynor, L. Brancaleon and H. Moseley, Photochem. Photobiol., 2004, 79, 105–113 CrossRef CAS PubMed
- H. Moseley, S. Ibbotson, J. Woods, L. Brancaleon, A. Lesar, C. Goodman and J. Ferguson, Lasers Surg. Med., 2006, 38, 403–416 CrossRef CAS PubMed
- M. Mozaffarieh, A. Schotzau, T. Josifova and J. Flammer, Mol. Vision, 2009, 15, 1194–1199 CAS
- S. Ohashi, O. Kikuchi, M. Tsurumaki, Y. Nakai, H. Kasai, T. Horimatsu, S. Miyamoto, A. Shimizu, T. Chiba and M. Muto, PLoS One, 2014, 9, e103126 Search PubMed
- C. S. Shemesh, C. W. Hardy, D. S. Yu, B. Fernandez and H. Zhang, Photodiagn. Photodyn. Ther., 2014, 11, 193–203 CrossRef CAS PubMed
- E. R. Blazek, J. G. Peak and M. J. Peak, Photochem. Photobiol., 1989, 49, 607–613 CrossRef CAS PubMed
- W. K. Pogozelski and T. D. Tullius, Chem. Rev., 1998, 98, 1089–1107 CrossRef CAS PubMed
- P. Prasad, I. Khan, P. Kondaiah and A. R. Chakravarty, Chem.–Eur. J., 2013, 19, 17445–17455 CrossRef CAS PubMed
- B. Banik, K. Somyajit, A. Hussain, G. Nagaraju and A. R. Chakravarty, Dalton Trans., 2014, 43, 1321–1331 RSC
- A. R. Chakravarty, J. Chem. Sci., 2006, 118, 443–453 CrossRef CAS
- D. Crespy, K. Landfester, U. S. Schubert and A. Schiller, Chem. Commun., 2010, 46, 6651–6662 RSC
- U. Schatzschneider, Eur. J. Inorg. Chem., 2010, 2010, 1451–1467 CrossRef
- A. R. Chakravarty and M. Roy, in Progress in Inorganic Chemistry, ed. K. D. Karlin, John Wiley & Sons, Inc, Hoboken, New Jersey, 2012, vol. 57, ch. 3, pp. 119–202 Search PubMed
- A. Hussain and A. R. Chakravarty, J. Chem. Sci., 2012, 124, 1327–1342 CrossRef CAS
- B. A. Armitage, Chem. Rev., 1998, 98, 1171–1200 CrossRef CAS PubMed
- T. Da Ros, G. Spalluto, A. S. Boutorine, R. V. Bensasson and M. Prato, Curr. Pharm. Des., 2001, 7, 1781–1821 CrossRef CAS PubMed
- L. Herman, S. Ghosh, E. Defrancq and A. K. D. Mesmaeker, J. Phys. Org. Chem., 2008, 21, 670–681 CrossRef CAS
- D. Magda, M. Wright, R. A. Miller, J. L. Sessler and P. I. Sansom, J. Am. Chem. Soc., 1995, 117, 3629–3630 CrossRef CAS
- S. Dhar, D. Senapati, P. A. N. Reddy, P. K. Das and A. R. Chakravarty, Chem. Commun., 2003, 2452–2453 RSC
- S. Dhar, D. Senapati, P. K. Das, P. Chattopadhyay, M. Nethaji and A. R. Chakravarty, J. Am. Chem. Soc., 2003, 125, 12118–12124 CrossRef CAS PubMed
- S. Dhar, M. Nethaji and A. R. Chakravarty, Dalton Trans., 2005, 344–348 RSC
- A. Fortner, S. G. Wang, G. K. Darbha, A. Ray, H. T. Yu, P. C. Ray, R. R. Kalluru, C. K. Kim, V. Rai and J. P. Singh, Chem. Phys. Lett., 2007, 434, 127–132 CrossRef CAS
- D. Lahiri, T. Bhowmick, B. Pathak, O. Shameema, A. K. Patra, S. Ramakumar and A. R. Chakravarty, Inorg. Chem., 2009, 48, 339–349 CrossRef CAS PubMed
- S. Roy, S. Saha, R. Majumdar, R. R. Dighe and A. R. Chakravarty, Inorg. Chem., 2009, 48, 9501–9509 CrossRef CAS PubMed
- P. K. Sasmal, A. K. Patra, M. Nethaji and A. R. Chakravarty, Inorg. Chem., 2007, 46, 11112–11121 CrossRef CAS PubMed
- P. K. Sasmal, S. Saha, R. Majumdar, R. R. Dighe and A. R. Chakravarty, Chem. Commun., 2009, 1703–1705 RSC
- P. Prasad, P. K. Sasmal, R. Majumdar, R. R. Dighe and A. R. Chakravarty, Inorg. Chim. Acta, 2010, 363, 2743–2751 CrossRef CAS
- P. K. Sasmal, S. Saha, R. Majumdar, R. R. Dighe and A. R. Chakravarty, Inorg. Chem., 2010, 49, 849–859 CrossRef CAS PubMed
- B. Banik, K. Somyajit, D. Koley, G. Nagaraju and A. R. Chakravarty, Inorg. Chim. Acta, 2012, 393, 284–293 CrossRef CAS
- S. Banerjee, A. Hussain, P. Prasad, I. Khan, B. Banik, P. Kondaiah and A. R. Chakravarty, Eur. J. Inorg. Chem., 2012, 3899–3908 CrossRef CAS
- B. Balaji, B. Banik, P. K. Sasmal, B. Maity, R. Majumdar, R. R. Dighe and A. R. Chakravarty, Eur. J. Inorg. Chem., 2012, 126–135 CrossRef CAS
- B. Banik, K. Somyajit, G. Nagaraju and A. R. Chakravarty, RSC Adv., 2014, 4, 40120–40131 RSC
- S. Banerjee, I. Pant, I. Khan, P. Prasad, A. Hussain, P. Kondaiah and A. R. Chakravarty, Dalton Trans., 2015, 44, 4108–4122 RSC
- B. Wilson, M. J. Fernandez, A. Lorente and K. B. Grant, Org. Biomol. Chem., 2008, 6, 4026–4035 CAS
- C. T. Mapp, E. A. Owens, M. Henary and K. B. Grant, Bioorg. Med. Chem. Lett., 2014, 24, 214–219 CrossRef CAS PubMed
- Y.-C. Zheng, M.-L. Zheng, K. Li, S. Chen, Z.-S. Zhao, X.-S. Wang and X.-M. Duan, RSC Adv., 2015, 5, 770–774 RSC
- J. L. Sessler and R. A. Miller, Biochem. Pharmacol., 2000, 59, 733–739 CrossRef CAS PubMed
- C. Preihs, J. F. Arambula, D. Magda, H. Jeong, D. Yoo, J. Cheon, Z. H. Siddik and J. L. Sessler, Inorg. Chem., 2013, 52, 12184–12192 CrossRef CAS PubMed
- K. Kadish, B. Maiya and C. Araullo-McAdams, J. Phys. Chem., 1991, 95, 427–431 CrossRef CAS
- M. F. Renschler, A. Yuen, T. J. Panella, T. J. Wiemann, S. Dougherty, L. Esserman, M. Panjehpour, S. W. Taber, V. H. Fingar, E. Lowe, J. S. Engel, B. Lum, K. W. Woodburn, W.-F. Cheong and R. A. Miller, Presented in part at the Optical Methods for Tumor Treatment and Detections: Mechanisms and Techniques in Photodynamic Therapy VII, San Jose, CA, May, 1998 Search PubMed
- K. Verigos, D. C. Stripp, R. Mick, T. C. Zhu, R. Whittington, D. Smith, A. Dimofte, J. Finlay, T. M. Busch, Z. A. Tochner, S. Malkowicz, E. Glatstein and S. M. Hahn, J. Environ. Pathol., Toxicol. Oncol., 2006, 25, 373–387 CrossRef CAS
- ClinicalTrials.gov, https://www.clinicaltrials.gov/ct2/results?term=lutex%26Search=Search, accessed October, 2015.
- L. Flowers, S. T. Ohnishi and T. M. Penning, Biochemistry, 1997, 36, 8640–8648 CrossRef CAS PubMed
- H. Speisky, M. Gomez, F. Burgos-Bravo, C. Lopez-Alarcon, C. Jullian, C. Olea-Azar and M. E. Aliaga, Bioorg. Med. Chem., 2009, 17, 1803–1810 CrossRef CAS PubMed
- H. Y. Chen, D. P. Xu, G. L. Tan, W. Cai, G. X. Zhang, W. Cui, J. Z. Wang, C. Long, Y. W. Sun, P. Yu, K. W. Tsim, Z. J. Zhang, Y. F. Han and Y. Q. Wang, J. Mol. Neurosci., 2015, 56, 977–987 CrossRef CAS PubMed
- S. U. Rehman, H. Zubair, T. Sarwar, M. A. Husain, H. M. Ishqi, S. Nehar and M. Tabish, Tumor Biol., 2015, 36, 1237–1244 CrossRef PubMed
- D. Pogocki and C. Schoneich, Free Radicals Biol. Med., 2001, 31, 98–107 CrossRef CAS PubMed
- A. Jain, N. K. Alvi, J. H. Parish and S. M. Hadi, Mutat. Res., 1996, 357, 83–88 CrossRef PubMed
- P. Bilski, C. Chignell, J. Szychlinski, A. Borkowski, E. Oleksy and K. Reszka, J. Am. Chem. Soc., 1992, 114, 549–556 CrossRef CAS
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1–21 CrossRef CAS
- D. E. Ryan, K. B. Grant and K. Nakanishi, Biochemistry, 1996, 35, 8640–8650 CrossRef CAS PubMed
- P. C. Wilkins, M. D. Johnson, A. A. Holder and D. C. Crans, Inorg. Chem., 2006, 45, 1471–1479 CrossRef CAS PubMed
- M. L. Macheda, S. Rogers and J. D. Best, J. Cell. Physiol., 2005, 202, 654–662 CrossRef CAS PubMed
- R. B. Hamanaka and N. S. Chandel, J. Exp. Med., 2012, 209, 211–215 CrossRef CAS PubMed
- P. Anand, A. B. Kunnumakkara, R. A. Newman and B. B. Aggarwal, Mol. Pharm., 2007, 4, 807–818 CrossRef CAS PubMed
- T. Atsumi, K. Tonosaki and S. Fujisawa, Anticancer Res., 2007, 27, 363–371 CAS
- D. Hanahan and R. A. Weinberg, Cell, 2000, 100, 57–70 CrossRef CAS PubMed
- D. Kessel and Y. Luo, J. Photochem. Photobiol., B, 1998, 42, 89–95 CrossRef CAS
- M. Lam, N. L. Oleinick and A. L. Nieminen, J. Biol. Chem., 2001, 276, 47379–47386 CrossRef CAS PubMed
- C. Andreini, I. Bertini, G. Cavallaro, G. L. Holliday and J. M. Thornton, J. Biol. Inorg. Chem., 2008, 13, 1205–1218 CrossRef CAS PubMed
- M. Valko, H. Morris and M. T. Cronin, Curr. Med. Chem., 2005, 12, 1161–1208 CrossRef CAS PubMed
- G. J. Fowler, R. C. Rees and R. Devonshire, Photochem. Photobiol., 1990, 52, 489–494 CrossRef CAS PubMed
- Y. Chen, W. Zheng, Y. Li, J. Zhong, J. Ji and P. Shen, Cancer Sci., 2008, 99, 2019–2027 CAS
- E. J. Lim, C. H. Oak, J. Heo and Y. H. Kim, Oncol. Rep., 2013, 30, 856–862 CAS
- H. J. Lim and C. H. Oh, Photodiagn. Photodyn. Ther., 2011, 8, 337–342 CrossRef CAS PubMed
- Q. T. Nguyen and R. Y. Tsien, Nat. Rev. Cancer, 2013, 13, 653–662 CrossRef CAS PubMed
- A. L. Vahrmeijer, M. Hutteman, J. R. van der Vorst, C. J. van de Velde and J. V. Frangioni, Nat. Rev. Clin. Oncol., 2013, 10, 507–518 CrossRef CAS PubMed
- L. S. Murakami, L. P. Ferreira, J. S. Santos, R. S. da Silva, A. Nomizo, V. A. Kuz'min and I. E. Borissevitch, Biochim. Biophys. Acta, 2015, 1850, 1150–1157 CrossRef CAS PubMed
- A. P. Gorka, R. R. Nani and M. J. Schnermann, Org. Biomol. Chem., 2015, 13, 7584–7598 CAS
- B. A. Armitage, in Top Heterocycl Chem, ed. L. Strekowski, Springer-Verlag Berlin Heidelberg, Berlin, Heidelberg, 2008, vol. 14, ch. 3, pp. 11–29 Search PubMed
- A. Gorman, J. Killoran, C. O'Shea, T. Kenna, W. M. Gallagher and D. F. O'Shea, J. Am. Chem. Soc., 2004, 126, 10619–10631 CrossRef CAS PubMed
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed. Engl., 2009, 48, 3244–3266 CrossRef CAS PubMed
- O. V. Przhonska, S. Webster, L. A. Padilha, H. Hu, A. D. Kachkovski, D. J. Hagan and E. W. Van Stryland, in Advanced Fluorescence Reporters in Chemistry and Biology I, Fundamentals and Molecular Design, ed. A. P. Demchenko, Springer Ser Fluoresc, Berlin, Heidelberg, 2010, vol. 8, pp. 105–148 Search PubMed
| This journal is © The Royal Society of Chemistry 2016 |